Background
Omenn syndrome (MIM 603554) is an autosomal recessive form of severe combined immunodeficiency (SCID) characterized by erythroderma, desquamation, alopecia, chronic diarrhea, failure to thrive, lymphadenopathy, and hepatosplenomegaly (see the image below).
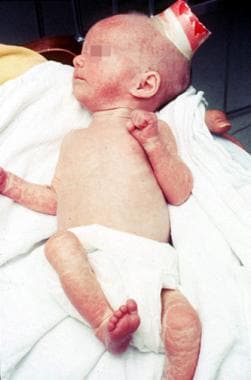 A unique dermatitis characterizes Omenn syndrome. The dermatitis initially resembles eczema, but with a pachydermia, as observed here. The lesions progress to desquamation. Failure to thrive is evident. This infant weighed 6 pounds at age 6 months; his weight had not changed since birth.
A unique dermatitis characterizes Omenn syndrome. The dermatitis initially resembles eczema, but with a pachydermia, as observed here. The lesions progress to desquamation. Failure to thrive is evident. This infant weighed 6 pounds at age 6 months; his weight had not changed since birth.
Cutaneous manifestations are common in primary immunodeficiency disorders. Erythroderma of infancy with diffuse alopecia is closely linked with children having SCID. [1] These patients may also develop fungal, bacterial, and viral infections typical of SCID. In this syndrome, the SCID is associated with the virtual absence of B cells and the presence of oligoclonal autoreactive T cells. [2] Lymphocytosis results from the expansion of an oligoclonal population of activated and antigen-stimulated T helper 2 (TH 2) cells that produce elevated levels of interleukin 4 (IL-4) and interleukin 5 (IL-5). The latter cytokines mediate eosinophilia and elevated immunoglobulin E (IgE) levels (see the image below).
This severe combined immunodeficiency disease has been linked to mutations in the RAG1/RAG2 gene. [3, 4]
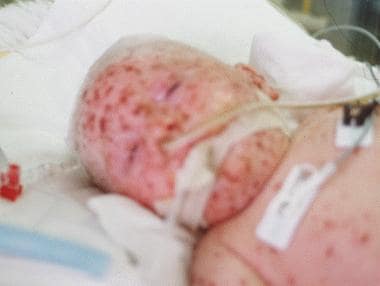 Common viral infections are fatal in severe combined immunodeficiency (SCID). This female infant died before bone marrow stem cell engraftment could occur, when varicella became resistant to acyclovir. The nasal bridge reveals superinfection with Klebsiella pneumoniae. Lymphedema, a characteristic of Omenn syndrome, is also shown.
Common viral infections are fatal in severe combined immunodeficiency (SCID). This female infant died before bone marrow stem cell engraftment could occur, when varicella became resistant to acyclovir. The nasal bridge reveals superinfection with Klebsiella pneumoniae. Lymphedema, a characteristic of Omenn syndrome, is also shown.
Pathophysiology
Impaired V(D)J recombination process leads to the generation of a few T cells expanding in the periphery, infiltrating target organs such as skin and gut, resulting in the erythroderma and colitis typical of this syndrome. [5] Thus, the inability to productively rearrange VDJ regions in T-cell and B-cell receptors leads to abnormal T cells and absent B cells.
The mutations in RAG-1 and RAG-2 in Omenn syndrome differ from T-cell negative (T-), B-cell negative (B-), and natural killer cell positive (NKC+) SCID caused by RAG-1 or RAG-2 mutations. In these conditions, the mutations affect the active core of the recombinase genes and typically negate the production of the recombinase protein; hence, no development of T-lineage and B-lineage cells occurs. In Omenn syndrome, the mutated RAG-1 and RAG-2 proteins remain normally distributed in the nucleus of cells. [6]
Early recognition of this condition is important for genetic counseling and early treatment. [7] The inflammation may be triggered by clonally expanded T cells, predominantly of the Th2 type. [8] These abnormal T cells presumably secrete cytokines that promote autoimmune as well as allergic inflammation. Omenn syndrome has been identified in leaky SCIDs caused by hypomorphic mutations in recombinase genes RAG-1 and RAG-2, which impair but do not eliminate recombination of variable, diversity, and joining (VDJ) segments of TCR and Ig genes. Most cases of Omenn syndrome reported so far are associated with hypomorphic mutations in RAG-1/RAG-2 genes.
A novel mechanism has been suggested: by selectively impairing recombination at certain coding flanks, a RAG mutant can cause primary repertoire restriction, as opposed to a more random, limited repertoire that develops secondary to severely diminished recombination activity, with autoimmune manifestations related to decreased thymic expression of tissue-specific antigens. [9]
However, Omenn syndrome is now known to occur in other leaky SCIDs with mutations in the RNA component of mitochondrial RNA processing endoribonuclease, adenosine deaminase, interleukin 2 (IL-2) receptor gamma, interleukin 7 (IL-7) receptor alpha, the nuclease ARTEMIS, and DNA ligase 4. Thus, Omenn syndrome is a distinct inflammatory process that can be associated with genetically diverse, leaky SCIDS. Accordingly, Omenn syndrome is best viewed, not as a specific form of SCID, but rather as an aberrant inflammatory condition that can be associated with multiple genetic abnormalities, which can significantly impair (but not abolish) T-cell development in the thymus. A novel homozygous frameshift mutation in IL7R (c.562delC) was indentified in the proband with both parents being Taiwanese aborigines who were delineated as carriers, none of whom had Omenn syndrome. [10] Mutations in the DCLRE1C gene, which encodes ARTEMIS, have been described in a number of patients. [11, 12] Many of the mutations were gross deletions of exons 1-3 or exons 1-4.
An oligoclonal expansion of Th2 population is viewed as a result of increased exposure to inadequately cleared antigens. These oligoclonal T cells have a highly restricted receptor repertoire, as well as increased apoptosis due to overexpression of CD95 and underexpression of anti-apoptotic factors, such as bcl -2.
Germinal centers are absent in the lymph nodes, which is consistent with the inability to produce functional antibodies. Hassall corpuscles are poorly formed, and lymphocytes are deficient in the thymus. Paracortical lymphocytes are absent in the spleen. Cutaneous Barrier Leakage may provide pivotal skin-gut interaction to sustain skin inflammation in OS. [13]
RAG -deficient mice have been developed. Their defects are restricted to the T- B- immunologic abnormalities, as observed in human RAG deficiency. Recently, 2 murine models bearing mutations of the VDJ recombinase analogous to those causing human OS have been developed. These murine models have oligoclonal T cells, an absence of circulating B cells, peripheral eosinophilia, and activated autoreactive T cells infiltrating gut and skin, causing diarrhea, alopecia, and, in some cases, severe erythrodermia. [14, 15]
Epidemiology
Frequency
The frequency of Omenn syndrome is difficult to ascertain. The prevalence of all forms of SCID is estimated to be 1 case per 50,000 population.
Omenn syndrome has been reported in patients from throughout the world, mainly North America and Europe.
Mortality/Morbidity
Omenn syndrome is fatal if untreated. Patients have life-threatening viral, bacterial, fungal, and Pneumocystis carinii infections that are observed in other types of SCID. Patients commonly have Staphylococcus aureus sepsis, which is related to the generalized dermatitis. Live viral infections, including those due to attenuated oral poliovirus, may cause death. In addition, chronic diarrhea and resulting inanition may be responsible for death.
Bone marrow transplantation (BMT) is usually successful, but life-threatening acute or chronic graft versus host disease (GVHD) may be a complication. This can occur in any stem cell reconstitution procedure.
Demographics
Patients have been identified in the United States, Canada, Europe, and India.
The incidences are equal among male and female infants; this observation is consistent with the autosomal recessive etiology of Omenn syndrome.
Infants present within weeks of birth and usually by age 3 months, as do those with other types of SCID. The characteristic dermatitis, chronic diarrhea, and failure to thrive often precede the onset of infections. Published reports of patients describe presentation by the time the patient is aged 6 months.
-
A unique dermatitis characterizes Omenn syndrome. The dermatitis initially resembles eczema, but with a pachydermia, as observed here. The lesions progress to desquamation. Failure to thrive is evident. This infant weighed 6 pounds at age 6 months; his weight had not changed since birth.
-
Common viral infections are fatal in severe combined immunodeficiency (SCID). This female infant died before bone marrow stem cell engraftment could occur, when varicella became resistant to acyclovir. The nasal bridge reveals superinfection with Klebsiella pneumoniae. Lymphedema, a characteristic of Omenn syndrome, is also shown.
Tables
Brand(Manufacturer) |
Manufacturing Process |
pH |
Additives (IVIG products containing sucrose are more often associated with renal dysfunction, acute renal failure, and osmotic nephrosis, particularly with preexisting risk factors [eg, history of renal insufficiency, diabetes mellitus, age >65 y, dehydration, sepsis, paraproteinemia, nephrotoxic drugs].) |
Parenteral Form and Final Concentrations |
IgA Content (mcg/mL) |
Carimune NF (CSL Behring) |
Kistler-Nitschmann fractionation; pH 4 incubation, nanofiltration |
6.4-6.8 |
6% solution: 10% sucrose, < 20 mg NaCl/g protein |
Lyophilized powder 3%, 6%, 9%, 12% |
Trace |
Flebogamma (Grifols USA) |
Cohn-Oncley fractionation, PEG precipitation, ion-exchange chromatography, pasteurization |
5.1-6 |
Sucrose free, contains 5% D-sorbitol |
Liquid 5% |
< 50 |
Gammagard Liquid 10% (Baxter Bioscience) |
Cohn-Oncley cold ethanol fractionation, cation and anion exchange chromatography, solvent detergent treated, nanofiltration, low pH incubation |
4.6-5.1 |
0.25M glycine |
Ready-for-use Liquid 10% |
37 |
Gamunex (Talecris Biotherapeutics) |
Cohn-Oncley fractionation, caprylate-chromatography purification, cloth and depth filtration, low pH incubation |
4-4.5 |
Contains no sugar, contains glycine |
Liquid 10% |
46 |
Gammaplex (Bio Products) |
Solvent/detergent treatment targeted to enveloped viruses; virus filtration using Pall Ultipor to remove small viruses including nonenveloped viruses; low pH incubation |
4.8-5.1 |
Contains sorbitol (40 mg/mL); do not administer if fructose intolerant |
Ready-for-use solution 5% |
< 10 |
Iveegam EN (Baxter Bioscience) |
Cohn-Oncley fraction II/III; ultrafiltration; pasteurization |
6.4-7.2 |
5% solution: 5% glucose, 0.3% NaCl |
Lyophilized powder 5% |
< 10 |
Polygam S/D Gammagard S/D (Baxter Bioscience for the American Red Cross) |
Cohn-Oncley cold ethanol fractionation, followed by ultracentrafiltration and ion exchange chromatography; solvent detergent treated |
6.4-7.2 |
5% solution: 0.3% albumin, 2.25% glycine, 2% glucose |
Lyophilized powder 5%, 10% |
< 1.6 (5% solution) |
Octagam (Octapharma USA) 9/24/10: Withdrawn from market because of unexplained reports of thromboembolic events |
Cohn-Oncley fraction II/III; ultrafiltration; low pH incubation; S/D treatment pasteurization |
5.1-6 |
10% maltose |
Liquid 5% |
200 |
Panglobulin (Swiss Red Cross for the American Red Cross) |
Kistler-Nitschmann fractionation; pH 4, trace pepsin, nanofiltration |
6.6 |
Per gram of IgG: 1.67 g sucrose, < 20 mg NaCl |
Lyophilized powder 3%, 6%, 9%, 12% |
720 |
Privigen Liquid 10% (CSL Behring) |
Cold ethanol fractionation, octanoic acid fractionation, and anion exchange chromatography; pH 4 incubation and depth filtration |
4.6-5 |
L-proline (approximately 250 mmol/L) as stabilizer; trace sodium; does not contain carbohydrate stabilizers (eg, sucrose, maltose) |
Ready-for use liquid 10% |
< 25 |




