Practice Essentials
Chronic granulomatous disease (CGD), an inherited disorder of phagocytic cells, results from an inability of phagocytes to produce bactericidal superoxide anions (O2-). [1, 2] This consequently interferes with the production of hydrogen peroxide (H2O2), hypochlorous acid (HOCI), and hydroxyl radicals (OH.), products that play a critical role in killing certain pathogenic bacterial and fungal agents. These deficits lead to recurrent, life-threatening bacterial and fungal infections. In addition, most patients with chronic granulomatous disease have dysregulated T helper (Th)-17 lymphocyte–controlled inflammation.
The nitroblue tetrazolium (NBT) and dihydrorhodamine (DHR) tests, as well as genetic testing, are indicated in the workup of chronic granulomatous disease. Antimicrobial prophylaxis, early and aggressive treatment of infections, and interferon-gamma are the cornerstones of current therapy for this disease.
Since its first description in the 1950s as a syndrome of recurrent infections, hypergammaglobulinemia, hepatosplenomegaly, and lymphadenopathy in males who invariably died in the first decade of life, notable advances have been made in the understanding of this disease. The outlook for affected patients has also improved.
Chronic granulomatous disease is now known to be caused by a defect in the nicotinamide adenine dinucleotide phosphate (NADPH), reduced form, oxidase enzyme complex of phagocytes. Chronic granulomatous disease refers to the characteristic granulomas that develop in response to chronic inflammation.
Although chronic granulomatous disease was once fatal in childhood, current preventive therapies and early detection of infectious complications allow 90% of children with the disorder to reach adulthood. [3]
Signs and symptoms of chronic granulomatous disease
The hallmark of CGD is early onset of severe, recurrent bacterial and fungal infections. Common presentations of the condition include the following:
-
Skin infections
-
Pneumonia
-
Lung abscesses
-
Suppurative lymphadenitis
-
Diarrhea secondary to enteritis
-
Perianal or perirectal abscesses
-
Hepatic or splenic abscesses
Another characteristic manifestation of CGD is the development of granulomas in the skin, gastrointestinal (GI) tract, and genitourinary (GU) tract.
Workup in chronic granulomatous disease
The standard assay for phagocytic oxidase activity is the NBT test, while the DHR test is now widely and commercially available and should be considered the preferred screening and diagnostic test for CGD. Testing for specific gene mutation is useful to establish the genetic inheritance pattern of CGD and aid in family counseling.
Imaging studies such as chest radiography and computed tomography (CT) scanning are valuable in the diagnosis and management of pulmonary and hepatosplenic infections.
The two most frequent findings on histologic examination of CGD lesions are infection and postinfectious granulomas.
Management of chronic granulomatous disease
Daily prophylaxis of bacterial infections with trimethoprim-sulfamethoxazole (TMP-SMZ; Bactrim) is indicated. Moreover, interferon-gamma is now recommended as life-long therapy for infection prophylaxis in CGD.
Patients with superficial or deep infections (vs those with obstructing granulomas) should receive aggressive antibiotics; the initial route is parenteral.
Hematopoietic stem cell transplantation (HSCT) is the only curative therapeutic modality currently available for CGD.
Despite the increased risk of wound healing associated with surgical intervention, surgery is still an important tool for patients with this disease. Operative treatment may be required to relieve obstruction of ureters from large granulomas, drainage of abscesses, and aggressive removal of established infection, especially in the lung and liver.
Pathophysiology
In response to phagocytosis, neutrophils normally increase their oxygen consumption, which has been termed the respiratory or oxidative burst. The clinical significance of the respiratory burst was made evident when neutrophils from patients with chronic granulomatous disease were shown to have a lack of increased oxygen consumption.
Chronic granulomatous disease is caused by a defect in phagocytic NADPH oxidase, which is responsible for producing O2-. This superoxide anion is then converted to relatively bactericidal reactive oxidants, such as hydroxyl radical (OH-), hydrogen peroxide (H2 O2), peroxynitrite anion (ONOO-), and oxyhalides (HOX-, in which the X moiety is most commonly chlorine). The superoxide anion is generated by transferring electrons from the reduced NADPH to molecular O2 in response to physiologic stimuli, such as phagocytosis. This reaction is mediated by the phagocyte NADPH oxidase otherwise known as phagocyte oxidase (phox).
Nitric oxide (NO) and other reactive nitrogen intermediates have a prominent microbicidal role in experimental animals but do not appear to have a critical role in human phagocytes.
The phox system is an NADPH oxidase enzyme complex consisting of 5 component proteins. Glycoprotein 91 (gp91) and protein 22 (p22) make up the b and a subunits of a membrane bound heterodimer referred to as flavocytochrome b558. Protein 47 (p47), protein (p67), and protein 40 (p40) exist together as the cytosolic components of phox. The membrane-bound (gp91 and p22) and cytosolic components (p47, p67, and p40) assemble at the phagolysosome membrane in response to inflammatory stimuli such as phagocytosis. The assembled enzyme complex transports electrons from cytosolic NADPH across the membrane to molecular oxygen inside the phagolysosome to generate superoxide and other more toxic radicals, such as hydrogen peroxide mediated by superoxide dismutase and HOX.
The precise mechanism by which this intracellular bleach kills microorganisms is still debated. Numerous additional cytosolic oxidase factors (rac1, rac2) and a membrane-associated factor, rap1A, have been identified as having important roles in oxidase activation and function. Chronic granulomatous disease results from defects in gp91, p22, p47, and p67. Thus far, no cases related to a defect in p40 have been reported. An immunodeficiency syndrome similar to chronic granulomatous disease was described in one patient secondary to a mutation involving rac2 (guanosine triphosphate [GTP]–bound signaling protein).
The most common molecular defect in chronic granulomatous disease is a mutation in the CYBB (cytochrome B, b subunit) gene that is located on the X chromosome and that encodes for gp91 (the b subunit of cytochrome b558). [4] The resulting syndrome is commonly called X-linked chronic granulomatous disease (X-CGD). Gp91 deficiency accounts for 50-70% of all cases of chronic granulomatous disease. More than 350 mutations in the CYBB gene are known, and thus far, all are unique to individual families. Data from analyses of carriers suggests that de novo mutations occur in about 10% of cases.
The second most common mutation occurs in the NCF1 gene on chromosome 7 that encodes for p47. This mutation is the most common autosomal recessive form of the disease, accounting for 20-40% of all cases of chronic granulomatous disease. Unlike CYBB which has more than 350 mutations, the NCF1 mutation is highly conserved to a single deletion in more than 90% of patients.
Mutations in the genes NCF2 (which encodes p67) and CYBA (which encodes p22) are rare, accounting for fewer than 10% of all cases of chronic granulomatous disease. Both of these mutations result in the autosomal recessive forms of chronic granulomatous disease.
About 95% of the mutations mentioned above result in complete absence or greatly diminished level of the affected protein. In the remaining 5%, a normal level of defective protein is produced. The 4 forms of the disease are referred to as X91 (X-linked, gp91), A22 (autosomal, p22), A47, and A67 CGD. The superscript+,-, oro is added to denote a normal level, a reduced level, or complete absence of the affected subunit.
Less than 10% of patients have the X-linked variant form of CGD (X91-), which has a relatively mild clinical course. Most of these patients have low but detectable levels of flavocytochrome b588, and their phagocytes can generate measurable amounts of superoxide. Defects in p47 also seem to be associated with enzymatic and clinical deficiency less profound than that observed in other forms. Diagnosis in adulthood is not uncommon in these patients with residual phox activity.
The chronic granulomatous disease phagocyte can kill numerous microorganisms despite its defects because most microorganisms endogenously produce hydrogen peroxide, which the chronic granulomatous disease–affected phagocyte can modify and use against the organism in the phagosome. Bacteria and fungi that cause most infections in chronic granulomatous disease are catalase-positive organisms. These microorganisms produce catalase that breaks down endogenously produced hydrogen peroxide; the generation of oxygen radicals by a normally functioning phox system is needed to ensure the death of these infecting microorganisms. [5]
Whereas both Pseudomonas aeruginosa and Burkholderia cepacia (also known as Pseudomonas cepacia) are catalase-positive organisms, the former is a rare pathogen in chronic granulomatous disease because chronic granulomatous disease neutrophils can kill P aeruginosa organisms by means of nonoxidative mechanisms. B cepacia is an important cause of infections in chronic granulomatous disease perhaps because of as-yet unexplained abilities to resist killing in neutrophil-mediated nonoxidative pathways. [6]
Fungal infections occur in as many as 20% of patients with chronic granulomatous disease. The most common pathogens are Aspergillus fumigatus, Torulopsis glabrata (ie, Candida glabrata), and Candida albicans.Pneumonia is the most common presentation of fungal infection. Aspergillus nidulans, which is a rare pathogen in other patient populations, has emerged as a problematic pathogen in chronic granulomatous disease. It causes locally invasive or disseminated disease that is more lethal than that caused by A fumigatus. In a review of a registry of patients with chronic granulomatous disease, Aspergillus infection was the leading cause of death (see the image below), and B cepacia infection was the second most common.
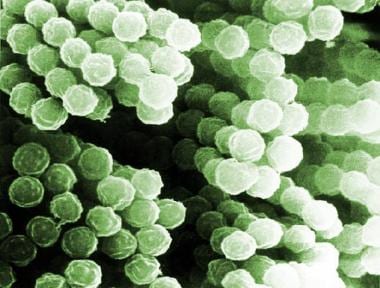 Scanning electron micrograph of Aspergillus species.
Scanning electron micrograph of Aspergillus species.
The diagnosis of chronic granulomatous disease should be considered in any patient with recurrent infections with catalase-positive organisms; infections with unusual organisms such as Serratia marcescens, A nidulans, or B cepacia; or infections in sites normally considered to be rare in children, such as a Staphylococcus aureus infection in a liver abscess. Sepsis is a common cause of death in CGD.
Epidemiology
Frequency
United States
The exact incidence of chronic granulomatous disease (CGD) is unknown. Analysis of data submitted to a national registry suggests that the incidence of CGD in the United States is about 1 case per 200,000-250,000 population (as many as 20 patients with CGD are born each year), with no apparent racial or ethnic predilection.
International
Surveys from the Netherlands and other parts of the world suggest a frequency of about 1 case per 220,000-500,000 population. [7]
Mortality/Morbidity
A detailed study of the natural history of CGD is unavailable. The registry data suggest that both morbidity and mortality rates are highest in patients with the X-linked form of the disease. A substantial number of patients in the registry died during the second and third decades of life, though some survived beyond the fourth decade. Approximately 80% of patients were alive at 5 years after they were entered in the registry. Even in the modern age of care for this disease, sporadic data suggest a potential excess in mortality in individuals aged 10-30 years. In a European study of 429 patients, based on 2000-2003 data, mean survival time for patients with X-linked CGD (gp91phox deficient) was 37.8 years, and for those with autosomal recessive CGD, 49.6 years. [8]
Race
No racial predilection is known.
Sex
About two thirds of cases are inherited as X-linked defects, and the remaining cases are inherited in autosomal recessive fashion. Of the 368 patients from 318 kindreds reported to the chronic granulomatous disease registry, 316 (86%) were male.
Age
Although the vast majority of affected individuals present with infections in early childhood, several reports describe affected patients who became symptomatic later than this. Chronic granulomatous disease is probably undiagnosed in some patients because they have a clinically mild phenotype.
-
Scanning electron micrograph of Aspergillus species.




