Practice Essentials
Nephrotic syndrome (or nephrosis, a term found in older literature and which does not capture the syndromic aspects of the disorder [1] ) is defined by the presence of nephrotic range proteinuria, hyperlipidemia, hypoalbuminemia, and edema. Nephrotic range proteinuria refers to a large amount of protein detected by the dipstick test (3+ or 4+) or by direct quantitative measurement. In children it is defined as protein excretion of more than 40 mg/m2 of body surface area/hr (referenced to body surface area to account for varying body sizes throughout childhood). A first-morning urine protein/creatinine ratio of 2-3 mg/mg creatinine or more indicates nephrotic range proteinuria. Since the dipstick detects albumin, proteinuria in the context of nephrotic syndrome is actually albuminuria.
Signs and symptoms
The insidious onset of edema is the presenting symptom in about 95% of children with nephrotic syndrome. Edema is typically first noted in the face and the lower eyelids (periorbital) and somewhat later in the lower extremities, scrotum or labia, and abdomen (ascites). Facial and periorbital edema are often more pronounced in the morning but dissipates later in the day as the edema settles to the lower part of the body. Clothes which formerly fit will now be tight. Socks wth an elastic band may leave an indentation. Marked edema of multiple body regions is anasarca. It is not uncommon for periorbital swelling to be mistaken for an allergic reaction by caretakers and primary care providers, until the progression of the edema or urine testing indicates the presence of proteinuria. Edema of the lower extremities or lower back is pitting.
Other signs and symptoms may include the following:
-
Viral respiratory tract infection: A history of a respiratory tract infection immediately preceding the onset of nephrotic syndrome is frequent on initial presentation and on subsequent relapses.
-
Allergy: Approximately 30% of children with nephrotic syndrome have a history of allergy. [2]
-
Microhematuria: Gross or macroscopic hematuria is rare and may indicate a complication such as infection or renal vein thrombosis.
-
Symptoms of infection: May include fever, lethargy, irritability, or abdominal pain due to sepsis or peritonitis.
-
Hypotension and signs of shock: Can be present in children presenting with sepsis or with marked hypovolemia.
-
Respiratory distress: Due to either massive ascites and pressure against the diaphragm or frank pulmonary edema, although pleural effusion is more likely than pulmonary congestion or edema.
-
Seizure: Caused by cerebral thrombosis.
-
Diminished appetite
-
Abdominal discomfort, pain, and peritoneal signs: Resulting from spontaneous bacterial peritonitis, ascites, or bowel wall edema. Occasionally presumed bowel wall edema causes abdominal pain with severe cramps and a clinical presentation resembling intussception. The presntation of an "acute abdomen" has resulted in a laporotomy for suspected acute appendicitis in instances when a urinalysis was not done.
-
Diarrhea: Due to bowel wall edema or malabsorption.
-
Hypertension: Resulting from fluid overload or primary kidney disease (unusual in minimal change disease).
See Presentation for more detail.
Diagnosis
In order to establish the presence of nephrotic syndrome, laboratory tests should confirm (1) nephrotic range proteinuria, (2) hypoalbuminemia, and (3) hyperlipidemia. Initial laboratory testing includes the following:
-
Urinalysis
-
Urine protein quantification (preferably first-morning urine protein-to-creatinine ratio)
-
Serum albumin measurement
-
Lipid panel
The following tests should be performed in selected patients to determine whether the nephrotic syndrome is primary and idiopathic (INS) or secondary and, if INS has been confirmed, whether signs of chronic kidney disease or extrarenal disease exclude minimal change nephrotic syndrome (MCNS):
-
Complete blood cell (CBC) count
-
Complete metabolic panel that includes serum electrolytes, calcium, phosphorus, albumin, blood urea nitrogen (BUN), creatinine, aspartate aminotransferase (AST), and alanine aminotransferase (ALT)
-
Ionized calcium (the total calcium level will be low due to hypoalbuminemia)
To exclude a secondary form of nephrotic syndrome in selected patients, the following tests may be considered:
-
Testing for human immunodeficiency virus (HIV)
-
Testing for hepatitis B and C viruses
-
Complement components (C3, C4)
-
Antinuclear antibody (ANA), anti–double-stranded DNA antibody (in selected patients)
Other tests and procedures in selected patients may include the following:
-
Genetic studies
-
Kidney ultrasonography
-
Chest radiography
-
Mantoux test or Quantiferon Gold TB test
-
Kidney biopsy
See Workup for more detail.
Management
Corticosteroids
If kidney biopsy is not initially indicated, a trial of corticosteroids is the first step in the treatment of INS if the clinical picture fits that of MCNS. See Guidelines for details about dose and duration of treatment.
Diuretics and albumin
Loop diuretics, such as furosemide, are often used to lessen edema. Metolazone (a thiazide diuretic inhibiting an ion transporter at a nephron site more distal to the loop) may be beneficial for edema that does not respond to furosemide alone. Intravenous 25% albumin can be combined with diuretics and may be useful in diuretic-resistant edema and in patients with significant ascites or scrotal, penile, or labial edema. Caution should be used when administering albumin; in addition to pulmonary edema and/or hypertension, albumin infusion may result in acute kidney injury and an allergic reaction. A different perspective is that albumin infusion may "overload" the proximal tubular reclamation process of albumin absorption, with the potential for tubular and interstitial injury as a consequence of the generation of reactive oxygen species (see discussion below regarding tubular injury as a consequence of unremitting heavy albuminuria).
Antihypertensive agents
Angiotensin-converting enzyme (ACE) inhibitors and angiotensin II receptor blockers (ARBs) can reduce hypertension and may also contribute to reducing proteinuria. However, because ACE inhibitors and ARBs can cause birth defects, adolescent females who are taking these agents must be counseled regarding the use of birth control, and pregnancy testing should be considered before starting these agents. A small increase in serum creatinine is common after initiation of the above medications as a consequence of efferent arteriolar vasodilation.
Calcium channel blockers and beta-blockers may also be used as first-line agents for hypertension.
Steroid-sparing agents
This group includes cyclophosphamide, calcineurin inhibitors, rituximab, and mycophenolate mofetil.
Cyclophosphamide (CYP)
CYP is often used in children with suspected or biopsy-proven MCNS with frequent relapses to sustain a corticosteroid-induced remission, but with the possible risks of infertility and other adverse effects. With the advent of newer agents, such as rituximab (an agent suppressing CD20 B lymphocytes), CYP is used less often for the management of steroid-dependent nephrotic syndrome.
Calcineurin inhibitors
Calcineurin inhibitors (eg, cyclosporin A [CSA], tacrolimus [TAC]) are most often used in patients with INS associated with the lesion of focal segmental glomerulosclerosis (FSGS)), or patients with suspected or biopsy-proven MCNS with frequent relapses who fail to respond to CYP or whose parents refuse other treatments (such as CYP). TAC is preferable but because of the risk of nephrotoxicity dosing must be monitored by means of trough tacrolimus blood levels.
Rituximab
Rituximab, a chimeric (IgG with both a mouse and human component) monoclonal antibody against CD20 that depletes B cells, has been used with increasing frequency in patients with suspected or biopsy-proven MCNS and frequent relapses who fail to respond to other steroid-sparing treatments.
Home monitoring
All patients and parents should be taught to monitor and maintain a record of first-morning urine protein at home with urine test strips. Urine testing at home is also useful in monitoring the response (or the lack of a response) to corticosteroid treatment. If a scale is available, body weight should be monitored and recorded under the same conditions each morning concomitant with the period of proteinuria. The advent of smart phones facilitates photography of facial edema, pitting edema of the lower extremities, color change on the test strip, and communication with the nephrologist.
See Treatment and Medication for more detail.
Background
Nephrotic syndrome is defined by nephrotic range proteinuria, hyperlipidemia, hypoalbuminemia, and edema. Nephrotic range proteinuria in adults is protein excretion of 3.5 g or more per day. However, because of the wide range of body sizes in children, there is not a single quantitative reference value for nephrotic range proteinuria. In children it is protein excretion of more than 40 mg/m2/hr. Since 24-hour urine collections may be unreliable and burdensome, especially in young children, a single, first-morning urine sample to quantify protein excretion by the ratio of protein to creatinine is preferable. [3] The use of a first-morning urine sample also eliminates the possibility of orthostatic proteinuria, which is not a pathological entity and might increase protein in a urine sample into the nephrotic range if collected while a patient is active during the day. A urine protein/creatinine ratio of more than 2-3 mg/mg indicates nephrotic range proteinuria and correlates with results from 24-hour urine collection. [4]
Nephrotic syndrome is the consequence of massive urinary losses of protein. Thus, nephrotic syndrome is not a disease itself, but the manifestation of several different glomerular disorders. These diseases might be acute and transient, such as postinfectious glomerulonephritis, or chronic and progressive, such as chronic kidney disease of diverse etiology associated with the glomerular lesion of focal segmental glomerulosclerosis (FSGS). Minimal change nephrotic syndrome (MCNS) often has a course of relapses of proteinuria and remissions. Nephrotic syndrome is a manifestation of a glomerular lesion rather than inflammation of the tubules or interstitium.
The glomerular diseases that cause nephrotic syndrome can be divided into primary and secondary etiologies. Primary nephrotic syndrome, also known as idiopathic nephrotic syndrome (INS), is associated with glomerular diseases intrinsic to the kidney and not related to systemic disorders. The subcategories of INS are based on histopathological descriptions. A variety of glomerular lesions can be seen in INS. These lesions include MCNS, FSGS, membranous nephropathy (MN), membranoproliferative glomerulonephritis (MPGN), C3 glomerulonephritis (C3GN), IgA nephropathy, and diffuse mesangial proliferation.
By definition, secondary nephrotic syndrome refers to an etiology extrinsic to the kidney. Among the many secondary causes of nephrotic syndrome are the following (which are less common in children than in adults):
-
Autoimmune and vasculitic diseases, such as Henoch-Schönlein purpura (HSP), systemic lupus erythematosus, and antineutrophil cytoplasmic antibody (ANCA)–associated vasculitis
-
Infectious diseases, such as congenital syphilis, malaria, human immunodeficiency virus (HIV) infection, hepatitis B and C, and in the current era, COVID-19
-
Malignancy
-
Environmental and drug exposure, such as heroin and mercury
-
An endocrine disorder; poorly controlled diabetes mellitus
-
An inborn error of metabolism, such as Fabry disease
Single gene (monogenic) disorders may cause nephrotic syndrome (NS). Congenital NS (presenting before age 3 mo) and infantile NS (presenting at age 4-12 mo) have been associated with defects in the nephrin gene (NPHS1), phospholipase C epsilon 1 gene (PLCE1), and the Wilms tumor suppressor gene (WT1). Mutations in the podocin gene (NPHS2) are associated with a familial, autosomal-recessive form of FSGS. Mutations in the α-actinin-4 gene (ACTN4) and the gene TRPC6 are associated with autosomal-dominant forms of familial FSGS.
Primary INS is quintessentially an intrinsic disorder of podocytes; ie, a podocytopathy. [5] More than 50 genes have been associated with the podocytopathy of nephrotic syndrome, and approximately 30% of children with steroid-resistant nephrotic syndrome may be found to have a single-gene cause of their disease. [6] Other genetic disorders with syndromic features have been associated with a podocytopathy, such as nail-patella syndrome, Pierson syndrome, and Schimke immuno-osseous dysplasia.
The response to steroid treatment in a patient with INS serves to delineate important clinical subtypes; steroid-sensitive (SSNS) and steroid-resistant nephrotic syndromes (SRNS). A response to steroids has a high correlation with minimal-change nephrotic syndrome (MCNS) and a favorable prognosis. An early multicenter landmark study of nephrotic syndrome in children, the International Study of Kidney Disease in Children found that the vast majority of preadolescent children with INS had MCNS on kidney biopsy. [7, 8] Whereas 90% of children with MCNS responded to corticosteroid treatment with remission of proteinuria, only 20% of children with the FSGS lesion responded to steroids.
This article focuses on primary (idiopathic) childhood nephrotic syndrome. A discussion of congenital and secondary nephrotic syndrome is beyond the scope of this article.
Pathophysiology
Proteinuria and the glomerular filtration barrier
INS in most children is intrinsically a disorder of the podocyte. The massive proteinuria represents a disruption of the glomerular filtration barrier, the trilaminar array of the inner endothelial cell layer, the noncellular collagen type IV glomerular basement membrane, and the outermost layer, the podocytes (which interfaces with the urinary space of Bowman's capsule). A podocyte arborizes as multiple main branches extending from the cell body, each then extending secondary branches with foot processes (pedicels). It is the unique structure of the slit diaphragm bridging the gap between the foot processes of adjacent podocytes which accounts for the high hydraulic conductivity for electrolytes and other small molecules and the restriction to the passage of larger molecules such as albumin in the healthy kidney. The slit diaphragm is maintained in health by the equipoise of several components of the podocyte cytoskeleton. Although the foot processes are coated with sialoprotein, which has a net negative charge, it is believed that the property of selectivity mainly resides in the sieving by molecular size rather than an electrostatic repulsion with albumin, which also has a net negative charge.
Nephrotic range albuminuria is a consequence of the disruption of the podocyte cytoskeleton which, in health, "splints" the foot processes. It is the collapse of the cytoskeleton and the retraction of foot processes that result in the increased permeability of the glomerular filtration barrier to albumin and the foot process effacement seen on electron microscopy. The reversibility of the disrupted cytoskeleton and reconstitution of discrete foot processes upon successful treatment of MCNS with corticosteroids is a unique biological phenomenon.
The high hydraulic conductivity of the glomerular filtration barrier means that a large load of albumin is presented to the permselectivity mechanism. The restriction of the passage of this load of albumin when the filtration barrier is intact implies that the slit diaphragm should become clogged by the albumin that does not get through the slits. This posed a conundrum to the late Nobel laureate Oliver Smithies in speculating as to what actually happens to all of that albumin. [9] Since clogging has not been shown to actually exist, it seems that albumin may not be dragged that deep into the filtration barrier or its passage is resisted by the negatively-charged glycocalyx of the endothelial cell fenestrations. [10] As hydraulic conductivity drives filtration deeper through the serial layers of the filtration barrier, the permselectivity for excluding albumin from the filtrate is greatest at the level of the slit diaphragm, based primarily on size selectivity.
The homeostatic equilibrium necessary for health is critically dependent on the competing imperatives of elimination and conservation. Relevant to this discussion and considering that the kidney is a highly perfused and a high filtration organ, the amount of albumin presented to the kidney and coursing through the glomerular capillaries in one day in an adult male is over 27,000 grams. Were it not for the highly selective sieving by an intact glomerular filtration barrier, the potential loss of albumin would be incompatible with life. This is the essence of the meaning of the word, barrier, in understanding the imperative to prevent the loss of albumin while at the same time maintaining a high rate of perfusion and filtration. However, even in health the restriction of passage of albumin across the filtration barrier is not 100%, and one estimate is that about 3.3 g/day of albumin are filtered and 99% is conserved by the healthy kidney. [11]
Proteinuria and hypoalbuminemia
Possible immune pathogenesis
Immune system
The hallmark of idiopathic nephrotic syndrome (INS) is massive proteinuria, leading to decreased circulating albumin levels. The initiating event that produces proteinuria as a consequence of podocyte injury remains unknown. However, strong evidence suggests that INS, at least in part, has an immune pathogenesis. Parenthetically, nephrotic syndrome may also occur in conditions not assocciated with a primary podocytopathy, such as Alport syndrome.
The effect of glucocorticoids on inducing remission in INS implicates the immune system, and Shalhoub proposed that dysfunction of T lymphocytes contributed to the pathogenesis of the condition. [12] Glucocorticoids, primarily acting through the nuclear factor kappa B (NF-κB) transcription pathway, have a variety of effects, including inhibiting cytokine production and inhibition of T-cell production and proliferation. A variety of studies provide further evidence of the role of T cells in INS. [13] Patients with INS in remission have alterations in the NF-κB pathway compared with healthy control subjects. NF-κB transcription is up-regulated in relapse of INS compared with remission. Additionally, nephrotic syndrome has been reported in patients with Hodgkin lymphoma, a T-cell disease. Other observations in INS include altered thymic regulation of T-cell differentiation and alterations in T-cell subsets in INS patients compared with healthy controls.
In addition to T cells, the reports of remission in INS after treatment with rituximab, an anti-CD20 monoclonal antibody which causes clonal depletion of B lymphocytes, implicate a role for B cells in the pathogenesis of INS. [14, 15, 16, 17, 18]
A circulating factor may also play a role in the development of proteinuria in INS as seen by the rapid development of proteinuria in the recurrence of nephrotic syndrome after kidney transplantation in some patients with FSGS, the improvement in nephrotic syndrome in such patients after treatment with plasmapheresis, and the experimental induction of proteinuria in animals by plasma from some patients with INS and FSGS. [19] The nature of this circulating factor is not known. Various cytokines and molecules have been implicated, including the following [20] :
-
Interleukin (IL)-2, IL-4, IL-12, IL-13, IL-15, IL-18
-
IL-2 receptor
-
Interferon-γ
-
Tumor growth factor (TGF)-β
-
Vascular permeability factor
-
NF-κB
-
Tumor necrosis factor (TNF)-α
Wei et al reported an association between circulating levels of soluble urokinase receptor (suPAR) and FSGS in children and adults. [21, 22] Treatment of FSGS with immunosuppressive medications led to lower levels of suPAR The decline in suPAR levels over 26 weeks of treatment was associated with a reduction in proteinuria. Thus, suPAR might affect glomerular permeability. [21] However, subsequent studies have yielded conflicting data regarding suPAR, and the role of suPAR in the pathogenesis of FSGS and other glomerular diseases remains unclear. [23, 24]
The association of allergic responses with nephrotic syndrome also implicates the immune system in INS. Nephrotic syndrome has occurred after allergic reactions to bee stings, fungi, poison ivy, ragweed, house dust, jellyfish stings, and cat fur. Food allergy might play a role in relapses of INS; a reduced-antigenic diet was associated with improved proteinuria and complete remission in one study. [25, 26]
Additionally, INS is 3-4 times more likely in children with human leukocyte antigen (HLA)-DR7. Steroid-sensitive INS has also been associated with HLA-B8 and the DQB1 gene of HLA-DQW2. A greater incidence of INS is also observed in children with atopy and HLA-B12. [27]
More recent data point to a paradigm shift about the role of the immune system in the pathogenesis of MCNS in some patients. The etiology of INS of the minimal lesion type (MCNS) has been a puzzle for decades. Hence, the characterization as “idiopathic” seemed appropriate. Ever since the implication of the immune system by Shalhoub (mentioned above) and the responsiveness to steroids and immunomodulators, there has been a general sense that lymphocytes are implicated in some unknown way. However, this sense seemed not to be supported by the absence of classic indicia of an immune pathogenesis, in view of negative immunoflorescence and the absence of dense deposits on electron microscopy.
However, more recent research now supports a putative antibody-mediated mechanism for the podocytopathy in a subset of patients, mostly children. [28] Circulating anti-nephrin antibodies were demonstrated in 18 (29%) of 62 patients with biopsy-confirmed MCNS. After remission was induced, there were reduced or absent circulating antibodies. A unique feature was the presence of subtle punctate staining for IgG by immunofluorescence, which colocalized with nephrin in the slit diaphragm. These findings suggest a novel understanding of the disorder as an autoimmune disease, at least in some patients, and that the level of circulating antibody correlates with disease activity. If further research confirms these data, then this subset need no longer be labeled "idiopathic." This finding also supports the indication for rituximab for its anti-CD20 B cell activity, and presumably suppression of the B lymphocyte clone generating the anti-nephrin autoantibody in selected patients.
The mechanism of autoantibody production remains to be clarified. One may speculate that a viremia uncovers a hidden epitope or alters the extracellular domain of the nephrin molecule and creates a neoantigen triggering antibody production by a newly activated B cell clone, essentially a pauci-immune phenomenon. The paradox is that nephrin is abundant but that the punctate deposits are scattered. Nevertheless, the challenge is for further research to explain an autoimmune disease in the absence of florid classic immune pathological indicators in biopsy specimens.
The interaction of the extracellular domain of nephrin molecules bridging the gap between foot processes of neighboring podocytes is critical for the sieving property of the slit diaphragm. If the autoimmune mechanism is at play, one possible speculation is that antibody disrupts this interaction and propagates a disturbance to elements of the actin cytoskeleton. It is the collapse of the latter, retraction of foot processes, and avulsion of the slit diaphragm that is evident as diffuse foot process effacement during active disease in INS. The fate of nephrin bound to autoantibody is not known. It does not appear that there is a subtle pathologic variant of the NPHS1 gene that confers a genetic susceptibility for antibody-induced dysfunction, since 62 patients from the NEPTUNE (Nephrotic Syndrome Study Network) cohort in this study had whole genome sequencing of 68 genes associated with nephrotic syndrome, including NPHS1, and were not found to have a genetic basis for nephrotic syndrome.
Podocyte biology and genetics
Perhaps the most exciting developments in understanding the pathophysiology of nephrotic syndrome have occurred in the area of podocyte biology. [29] In this view, MCNS is a podocyopathy. The glomerular filtration barrier consists of the fenestrated capillary endothelium, the extracellular basement membrane, and the interdigitated podocyte foot processes between adjacent cells, connected by 200 nm slit diaphragms (see Figure). Nephrotic syndrome is associated with the biopsy finding of effacement of discrete interdigitating podocyte foot processes.
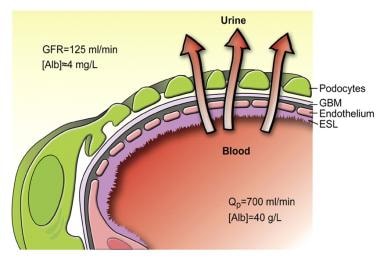 Schematic drawing of the glomerular barrier. Podo = podocytes; GBM = glomerular basement membrane; Endo = fenestrated endothelial cells; ESL = endothelial cell surface layer (often referred to as the glycocalyx). Primary urine is formed through the filtration of plasma fluid across the glomerular barrier (arrows); in humans, the glomerular filtration rate (GFR) is 125 mL/min. The plasma flow rate (Qp) is close to 700 mL/min, with the filtration fraction being 20%. The concentration of albumin in serum is 40 g/L, while the estimated concentration of albumin in primary urine is 4 mg/L, or 0.1% of its concentration in plasma. Courtesy of the American Physiological Society (www.the-aps.org) [Haraldsson B, Nystrom J, Deen WM. Properties of the glomerular barrier and mechanisms of proteinuria. Physiol Rev. 2008 Apr;88(2):451-87.]
Schematic drawing of the glomerular barrier. Podo = podocytes; GBM = glomerular basement membrane; Endo = fenestrated endothelial cells; ESL = endothelial cell surface layer (often referred to as the glycocalyx). Primary urine is formed through the filtration of plasma fluid across the glomerular barrier (arrows); in humans, the glomerular filtration rate (GFR) is 125 mL/min. The plasma flow rate (Qp) is close to 700 mL/min, with the filtration fraction being 20%. The concentration of albumin in serum is 40 g/L, while the estimated concentration of albumin in primary urine is 4 mg/L, or 0.1% of its concentration in plasma. Courtesy of the American Physiological Society (www.the-aps.org) [Haraldsson B, Nystrom J, Deen WM. Properties of the glomerular barrier and mechanisms of proteinuria. Physiol Rev. 2008 Apr;88(2):451-87.]
The current consensus is that injury or dysfunction of podocytes plays a primary role in the development of proteinuria. Insights into the molecular biology of the podocyte have greatly expanded the understanding of the pathophysiology of proteinuria in renal diseases.
Genetic associations with nephrotic syndrome
Various forms of INS have been described with genetic mutations affecting the podocyte, such as those associated with the following [30, 31] :
-
Slit-diaphragm and podocyte cytoskeleton: NPHS1, NPHS2, TRCP6, CD2AP, ACTN4, INF2, MYH9,MYO1E
-
Phospholipases and second-messenger systems: PLCE1
-
Glomerular basement membrane: LAMB2
-
Transcription factors: WT1, LMX1B
-
Lysosomal proteins: SCARB2
-
Mitochondrial proteins: COQ2
-
DNA-nucleosome restructuring mediator: SMARCAL1
Nephrin is a transmembrane protein that is a major structural element of the slit diaphragm and is encoded by the NPHS1 gene on chromosome 19. Mutations in the NPHS1 gene are responsible for autosomal recessive congenital nephrotic syndrome of the Finnish type (FCNS). FCNS is characterized by massive proteinuria in the first year of life (usually within the first 3 months) and progression to end-stage kidney disease within the first decade of life, although milder forms of the disease have been described. [30] Although mutations in NPHS1 are usually associated with congenital nephrotic syndrome, Philippe et al have described NPHS1 mutations in children aged 6 months to 8 years with later-onset steroid-resistant nephrotic syndrome (SRNS). [32] Santin et al have also described NPHS1 mutations in patients with later childhood-onset as well as adult-onset SRNS, providing the rationale for genetic testing in the instance of SRNS. [33]
Podocin is another podocyte protein that interacts with nephrin and is integral to the assembly of the slit diaphragm. Podocin is encoded by the NPHS2 gene on chromosome 1. Mutations in the NPHS2 gene were originally described in patients with autosomal recessive, steroid-resistant INS with FSGS on biopsy. Podocin mutations account for approximately 45-55% of familial and 8-20% of sporadic cases of SRNS. [30]
α-Actinin-4, encoded by the gene ACTN4 on chromosome 19, cross-links actin filaments of the podocyte cytoskeleton and anchors them to the glomerular basement membrane. The TRPC6 gene on chromosome 11 encodes a calcium channel associated with the slit diaphragm. [30] Disruptions in either ACTN4 or TRPC6 are associated with autosomal dominant forms of FSGS. [27]
CD2AP, which codes for a podocyte protein that associates with podocin and nephrin, has been linked to the development of nephrotic syndrome in animal models. However, the role it plays in human nephrotic syndrome is unclear. Various case reports have demonstrated heterozygous mutations in CD2AP in patients with nephrotic syndrome and FSGS. One report describes a single patient with a homozygous mutation in CD2AP and early onset of nephrotic syndrome with FSGS and diffuse mesangial sclerosis. [30]
Because African Americans have a 3- to 4-fold higher risk of end-stage kidney disease compared with persons of European ancestry, genetic studies have sought to explain this greater propensity to chronic kidney disease. A strong association was found in African Americans between idiopathic and HIV-related FSGS, as well as hypertensive end-stage kidney disease and mutations in the nonmuscle myosin heavy chain 9 (MYH9) gene. Nonmuscle MYH9 is a podocyte protein that binds to the podocyte actin cytoskeleton to perform intracellular motor functions. [34]
More recent studies have demonstrated that the increased risk of kidney disease previously ascribed to MYH9 is, in fact, more strongly associated with variations in the neighboring apolipoprotein L1 (APOL1) gene. Interestingly, these APOL1 variants, which are more common in African Americans but absent in whites, enhance the ability to lyse trypanosomes and may confer resistance to African sleeping sickness (Trypanosoma brucei rhodesiense infection). [35] Another nonmuscle myosin gene, MYO1E, was reported to be associated with FSGS in children. Mutation of the MYO1E gene led to disruption of the podocyte cytoskeleton. [36]
Other genetic forms of nephrotic syndrome continue to shed light on the pathogenesis of some forms of steroid-resistant INS. Mutations in the developmental regulatory gene WT1 are associated with congenital nephrotic syndrome associated with male pseudohermaphroditism, Wilms tumor (Denys-Drash syndrome), and gonadoblastoma (Frasier syndrome).
Mutations in phospholipase C epsilon 1 (PLCE1), a cytoplasmic enzyme required for podocyte maturation, have been associated with as many as 28% of cases of congenital nephrotic syndrome due to isolated (nonsyndromic) diffuse mesangial sclerosis. Nail-patella syndrome, a disorder characterized by skeletal and nail dysplasia as well as nephrotic syndrome, is caused by mutations in the LMX1B gene, which regulates expression of type IV collagen and the podocyte proteins nephrin, podocin, and CD2AP. [37]
Pierson syndrome, characterized by microcoria, abnormal lens shape, cataracts, blindness, severe neurologic deficits, congenital nephrotic syndrome, and progressive kidney failure, is caused by a mutation in the LAMB2 gene that codes for laminin b2, which is found in glomerular basement membrane, retina, lens, and neuromuscular synapses. [30]
Other rare forms of nephrotic syndrome with extrarenal manifestations have been associated with mutations in SCARB2, which codes for a lysosomal protein; disruption of this gene causes a syndrome of myoclonus epilepsy and glomerulosclerosis. Alterations in the mitochondrial protein coded by the gene COQ2 are associated with a syndrome of encephalopathy and nephropathy. Finally, mutations in the DNA-nucleosome restructuring mediator SMARCAL1 cause Schimke immuno-osseous dysplasia, a syndrome characterized by spondyloepiphyseal dysplasia (SED) resulting in disproportionate short stature, nephropathy, and T-cell deficiency. [31]
Monogenic causes of INS primarily result in SRNS. More than 39 genes have been associated with SRNS, and approximately 30% of children with SRNS may be found to have a single-gene cause of their disease. [6]
The role of podocyte gene alterations in minimal change nephrotic syndrome (MCNS) is unclear. Podocin appears to be expressed normally in MCNS but is decreased in FSGS. Mutations in nephrin and podocin do not appear to play a role in steroid-sensitive nephrotic syndrome. However, acquired alterations in slit diaphragm architecture might play a role in INS apart from actual mutations in the genes encoding podocyte proteins. Various authors have reported changes in expression and distribution of nephrin in MCNS.
Nongenetic associations with podocyte dysfunction
Coward et al demonstrated that nephrotic plasma induces translocation of the slit diaphragm proteins nephrin, podocin, and CD2AP away from the plasma membrane into the cytoplasm of the podocyte. [38] These authors also demonstrated that normal plasma might contain factors that maintain the integrity of slit diaphragm architecture and that the lack of certain factors (rather than the presence of an abnormal circulating factor) might be responsible for alterations in the podocyte architecture and the development of INS.
CD80, a T-cell costimulatory transmembrane protein, is expressed in podocytes and has been implicated in the pathogenesis of MCNS. Urinary CD80 levels are higher in patients with MCNS than in controls and patients with other glomerular diseases such as FSGS. Binding of interleukins or microbial products to toll-like receptors on the surface of the podocyte may lead to overexpression of CD80, as well as another protein, C-mip. CD80 and C-mip, in turn, may interfere with the proteins Nck and Fyn, leading to dephosphorylation of nephrin and disruption of the podocyte actin cytoskeleton, which result in conformational changes in the podocyte and slit diaphragm that cause proteinuria. [39] Blockade of CD80 by abatacept and belatacept has not been shown to attenuate proteinuria, however. [40] Hemopexin, a glycoprotein synthesized by the liver, may also induce nephrin-dependent changes in the podocyte skeleton that lead to proteinuria. [39] Apart from the podocyte and slit diaphragm, alterations in the glomerular basement membrane also likely play a role in the proteinuria of nephrotic syndrome (Alport syndrome).
In INS, the glomerular capillary permeability to albumin is increased, and this increase in filtered load exceeds the limited capacity of the tubules to reabsorb albumin from the filtrate. In its normal state, the glomerular basement membrane is negatively charged because of the presence of various polyanions along its surface, such as heparan sulfate, chondroitin sulfate, and sialic acid. This negative charge acts as a deterrent to filtration of negatively charged proteins, such as albumin. Experimental models in which the negative charges are removed from the basement membrane show an increase in albuminuria. Children with MCNS have been reported to have decreased anionic charges in the glomerular basement membrane. [37] Angiopoietin-like 4 and IL-8 may play a role in reducing anionic charges in the glomerular basement membrane. [39] Nevertheless a consensus has emerged that selectivity of sieving resides mainly in the property of molecular size rather than charge.
Edema
The classic explanation for edema formation is a decrease in plasma oncotic pressure, as a consequence of low serum albumin levels, causing an extravasation of plasma water into the interstitial space. The resulting contraction in plasma volume (PV) leads to stimulation of the renin-angiotensin-aldosterone axis and antidiuretic hormone. The resultant retention of sodium and water by the renal tubules contributes to the initiation and maintenance of edema.
Although this classic model of edema (also known as the "underfill hypothesis") seems logical, certain clinical and experimental observations do not completely support this traditional concept. First, the PV has not always been found to be decreased and, in fact, in most adults, measurements of PV have shown it to be increased. Only in young children with MCNS have most (but not all) studies demonstrated a reduced PV. Additionally, most studies have failed to document elevated levels of renin, angiotensin, or aldosterone—even during times of avid sodium retention. Active sodium reabsorption also continues despite actions that should suppress renin effects (eg, albumin infusion or angiotensin-converting enzyme [ACE] inhibitor administration).
Coupled with these discrepancies is the fact that, in the patient with steroid-responsive nephrotic syndrome, diuresis usually begins before the plasma albumin level has significantly increased and before the plasma oncotic pressure has changed. Some investigators have demonstrated a blunted responsiveness to atrial natriuretic peptide (ANP) despite higher than normal circulating plasma levels of ANP. [41]
Another model of edema formation, the "overfill hypothesis," postulates a primary defect in renal sodium handling. A primary increase in renal sodium reabsorption leads to net salt and water retention and subsequent hypertension. ANP might play a role in this mechanism; studies have shown an impaired response to ANP in nephrotic syndrome. This ANP resistance, in part, might be caused by overactive efferent sympathetic nervous activity, as well as enhanced tubular breakdown of cyclic guanosine monophosphate.
Other mechanisms that contribute to a primary increase in renal sodium retention include overactivity of the basolateral membrane ion transporter, Na+ -K+ -ATPase and the renal epithelial sodium channel (ENaC) in the cortical collecting duct and the shift of the Na+/H+ exchanger 3 (NHE3) from the inactive to active pools in the proximal tubule. [41]
A more recent theory of edema formation posits that massive proteinuria leads to tubulointerstitial inflammation and release of local vasoconstrictors and inhibition of vasodilation. Vasoconstriction of the afferent arteriole leads to a reduction in single-nephron glomerular filtration rate and sodium and water retention. [41]
Thus, the precise cause of edema and its persistence is uncertain. A complex interplay of various physiologic factors, such as the following, probably contribute:
-
Decreased oncotic pressure
-
Increased activity of aldosterone and vasopressin
-
Diminished ANP level
-
Activities of various cytokines and physical factors within the vasa recti
Hyperlipidemia
INS is accompanied by disordered lipid metabolism. Apolipoprotein (apo)-B–containing lipoprotein levels are elevated, including very-low-density lipoprotein (VLDL), intermediate-density lipoprotein (IDL), low-density lipoprotein (LDL), and lipoprotein(a), with resultant increases in total cholesterol and LDL-cholesterol. The level of high-density lipoprotein (HDL) cholesterol is normal or low. Elevations in triglyceride levels occur with severe hypoalbuminemia.
The traditional explanation for hyperlipidemia in INS was the increased synthesis of lipoproteins that accompany increased hepatic albumin synthesis due to hypoalbuminemia. However, serum cholesterol levels have been shown to be independent of albumin synthesis rates.
Decreased plasma oncotic pressure may play a role in increased hepatic lipoprotein synthesis, as demonstrated by the reduction of hyperlipidemia in patients with INS receiving either albumin or dextran infusions. Also contributing to the dyslipidemia of INS are abnormalities in regulatory enzymes, such as lecithin-cholesterol acyltransferase, lipoprotein lipase, and cholesterol ester transfer protein. [41, 42]
Thrombosis
Patients with nephrotic syndrome are at increased risk for thrombosis. The incidence of thromboembolic complications (TEC) is about 25% in adults with nephrotic syndrome.
The risk of TEC varies with the underlying disease. The incidence of TEC in infants with congenital nephrotic syndrome is about 10%. The risk of thrombosis increases throughout childhood, and adolescents are at higher risk than younger children after the first year of life. The risk of TEC also is greater in secondary than in primary nephrotic syndrome. Children with membranous nephropathy and nephrotic syndrome are at high risk for TEC, with an incidence of approximately 25%. [43] Zaffanello and Franchini found the subclinical rate of pulmonary embolism in children with nephrotic syndrome to be 28% using scintigraphic pulmonary ventilation and perfusion studies. [44]
The risk of TEC is greatest earlier in the course of nephrotic syndrome. The median time from diagnosis of nephrotic syndrome to TEC was 70 days in one study. Other studies have shown that the majority of TEC occur within the first 3 months of diagnosis. [43]
Renal vein thrombosis, deep vein thrombosis, and pulmonary embolism (PE) are the most frequently encountered TEC in children. Other venous sites of thrombosis include the superior sagittal sinus, other cerebral venous sites, and the inferior vena cava.
Arterial thrombosis, although less common than venous TEC, can occur and has been reported affecting the axillary, subclavian, femoral, coronary, and mesenteric arteries. [45]
Nephrotic syndrome is a hypercoagulable state. The increased risk of thrombosis can be attributed to 2 basic mechanisms: (1) urinary losses of antithrombotic proteins and (2) increased synthesis of prothrombotic factors. [46]
Decreased antithrombotic factors include the following:
-
Antithrombin III
-
Proteins C and S (conflicting data)
Increased synthesis of prothrombotic factors include the following:
-
Increased platelet number, activation, and aggregation
-
Elevation in levels of factors V and VIII, von Willebrand factor, α2-plasmin inhibitor, plasminogen activator inhibitor 1, and fibrinogen
-
Increased activities of tissue plasminogen activator and plasminogen activator inhibitor-1
These abnormalities in hemostatic factors, combined with potential hypovolemia, immobility, and increased incidence of infection, lead to a hypercoagulable state in INS. [2, 47]
Infection
Patients with INS are at increased risk for infection. Peritonitis and sepsis are the most common and serious infections. Peritonitis occurs at a rate of approximately 2-6% and may be accompanied by sepsis or bacteremia. The predominant bacterial causes are Streptococcus pneumoniae and gram-negative enteric organisms such as Escherichia coli. [48]
Other infections can also occur, including meningitis, cellulitis, viral infections, and others. Varicella is a particular concern in immunosuppressed patients and can be fatal. Prompt recognition and treatment with acyclovir (or postexposure prophylaxis with varicella-zoster immune globulin [VZIG]) is essential. Routine childhood varicella immunization has diminished some of the concern regarding this complication.
Infection, viral or bacterial, can trigger a relapse of INS and further complicate the course of the condition. The mechanism by which this occurs is not known.
Risk of infection may be increased in INS because of low immunoglobulin (Ig) G levels, which do not appear to be the result of urinary losses. Instead, low IgG levels seem to be the result of impaired synthesis, again pointing to a primary disorder in lymphocyte regulation in INS.
Additionally, increased urinary losses of factor B are noted. This is a cofactor of C3b in the alternative pathway of complement, which plays an important role in the opsonization of encapsulated organisms such as S pneumoniae. Impaired T-cell function may also be present in INS, which contributes to the susceptibility to infection. Finally, the medications used to treat INS, such as corticosteroids and alkylating agents, further suppress the immune system and increase the risk of infection. [2]
Acute kidney failure
Acute kidney failure (AKF) is a rare complication of INS, occurring in about 0.8% of cases. [49] Causes include the following [49] :
-
Rapid progression of underlying disease (nephrotic syndrome other than MCNS, secondary nephrotic syndrome) associated with this FSGS lesion
-
Bilateral renal vein thrombosis
-
Acute interstitial nephritis (AIN) due to drug therapy (eg, antibiotics, nonsteroidal anti-inflammatory agents [NSAIDs], diuretics)
-
Acute kidney injury (AKI) due to hypovolemia or sepsis
Use of ACE inhibitors or angiotensin II receptor blockers (ARBs) in conjunction with volume depletion can also precipitate AKF.
Etiology
Causes of INS include the following:
-
C3 glomerulonephritis
-
Idiopathic crescentic glomerulonephritis
Causes of genetic or congenital nephrotic syndrome include the following:
-
Finnish-type congenital nephrotic syndrome (NPHS1, nephrin)
-
Denys-Drash syndrome (WT1)
-
Frasier syndrome (WT1)
-
Diffuse mesangial sclerosis (WT1, PLCE1)
-
Autosomal recessive, familial FSGS (NPHS2, podocin)
-
Autosomal dominant, familial FSGS (ACTN4, α-actinin-4, TRPC6)
-
Nail-patella syndrome (LMX1B)
-
Pierson syndrome (LAMB2)
-
Schimke immuno-osseous dysplasia (SMARCAL1)
-
Galloway-Mowat syndrome
Infections that can cause secondary nephrotic syndrome include the following:
-
Congenital syphilis, toxoplasmosis, cytomegalovirus infection, rubella
-
Hepatitis B and C
-
HIV infection/acquired immunodeficiency syndrome (AIDS)
Drugs that can cause secondary nephrotic syndrome include the following:
-
Gold
-
Nonsteroidal anti-inflammatory drugs (NSAIDs)
-
Interferon
-
Mercury
-
Heroin
Systemic diseases that can cause secondary nephrotic syndrome include the following:
-
Systemic lupus erythematosus
-
Malignancy: Lymphoma, leukemia
-
Vasculitis: granulomatosis with polyangiitis (formerly known as Wegener granulomatosis), Churg-Strauss syndrome (eosinophilic granulomatosis with polyangiitis), polyarteritis nodosa, microscopic polyangiitis, Henoch-Schönlein purpura (HSP)
-
Immune-complex–mediated: Poststreptococcal (postinfectious) glomerulonephritis
Epidemiology
Incidence
In the United States, the reported annual incidence of nephrotic syndrome is 2-7 cases per 100,000 children younger than 16 years. The cumulative prevalence is approximately 16 cases per 100,000 individuals. [50] The International Study of Kidney Disease in Children (ISKDC) found that 76.6% of children with INS had MCNS on kidney biopsy findings, with 7% of cases associated with FSGS. [7, 51]
A study from New Zealand found the incidence of nephrotic syndrome to be almost 20 cases per million children under age 15 years. [52] In specific populations, such as those of Finnish or Mennonite origin, congenital nephrotic syndrome may occur in 1 in 10,000 or 1 in 500 births, respectively. [53]
Some studies have suggested a change in the histopathology of INS over the past few decades, although the overall incidence of INS has remained stable. The frequency of FSGS associated with INS appears to be increasing. A review of the literature suggested a 2-fold increase in the incidence of FSGS in recent decades. [54] However, another study found no evidence of an increasing incidence of FSGS. [55]
Race-, sex-, and age-related demographics
Black and Hispanic children appear to have an increased risk of steroid-resistant nephrotic syndrome and FSGS. [55, 56] An increased incidence of INS is reported in Asian children (6 times the rate seen in European children). An increased incidence of INS is also seen in Japanese and Southwest Asian children.
Primary steroid-sensitive nephrotic syndrome (SSNS) is rare in Africa, where nephrotic syndrome is more likely to be secondary or steroid-resistant. These variations in ethnic and geographic distribution of INS underscore the genetic and environmental influences in the development of PNS. [2]
In children younger than 8 years at onset, the ratio of males to females varies from 2:1 to 3:2 in various studies. In older children, adolescents, and adults, the male-to-female prevalence is approximately equal. ISKDC data indicate that 66% of patients with either MCNS or FSGS are male, whereas 65% of individuals with MPGN are female.
Of patients with MCNS, 70% are younger than 5 years. Only 20-30% of adolescents with INS have MCNS on biopsy findings. In the first year of life, genetic forms of INS and secondary nephrotic syndrome due to congenital infection predominate. [50]
Prognosis
Since the introduction of corticosteroids, the overall mortality of INS has decreased dramatically from over 50% to approximately 2-5%. Despite the improvement in survival, INS is often a chronic, relapsing disease and most patients experience some degree of morbidity, including the following:
-
Hospitalization, in some instances.
-
The burden of frequent monitoring both by parents and by physicians.
-
Administration of medications associated with significant adverse events.
-
A high rate of recurrence (relapses in >60% of patients).
-
The potential for progression to chronic kidney disease and end-stage kidney failure.
Additionally, INS is associated with an increased risk of multiple complications, including edema, infection, thrombosis, hyperlipidemia, acute kidney failure, and possible increased risk of cardiovascular disease.
The prognosis varies, depending on whether the nephrotic syndrome is steroid responsive or steroid resistant.
Steroid-responsive nephrotic syndrome
Patients who remain responsive to steroids with remission of proteinuria, even with frequent relapses, generally have a good prognosis. The ISKDC IInternational Study of Kidneyh Disease in Children) found that in 93% of children with INS who responded to steroids, kidney biopsy revealed MCNS. [8] In contrast, 75% of patients who did not initially respond to steroids had histology other than MCNS.
About 90% of children with MCNS (but only 20% of children with FSGS) achieve remission after the initial course of steroid treatment.
Despite the generally favorable prognosis in patients who respond to steroids, the ISKDC reported a 60% rate of subsequent relapses, which can lead to complications, increased morbidity, and decreased quality of life. [8] A longer course of initial steroid treatment (12 weeks rather than the original ISKDC protocol of 8 weeks) may reduce the rate of subsequent relapse to 36%, [57] which still represents a large number of patients who undergo repeated courses of immunosuppression, with possible hospitalizations, edema, infections, medication adverse effects, and other comorbidities.
A long-term study of 398 children with INS found that the percentage of children who became free of relapses during the course of their disease rose from 44% at 1 year after diagnosis to 69% at 5 years and 84% at 10 years after diagnosis. [50, 58] Although most children with INS who respond to steroids achieve long-term remission, relapses may continue into adulthood.
Older studies suggested that more than 90% of children achieve long-term remission without further relapses by puberty. However, this has been challenged by surveys indicating a rate of relapse during adulthood as high as 27-42%. [59]
A delay in the initiation of steroid treatment was associated with a worse prognosis in a seminal study by Heymann and Hunter. [60]
In a retrospective study, Vivarelli et al reported that the length of time between initiation of steroid treatment and remission is an early prognostic indicator for children with INS. [61] In study participants who did not suffer relapse or who relapsed infrequently, the median time from treatment onset to remission was less than 7 days. In patients who had frequent relapses or who developed steroid-dependent nephrotic syndrome, the median time to remission was more than 7 days.
A study of 42 adult patients with a history of childhood INS found that 33% of patients continued to relapse into adulthood. Fortunately, overall morbidity (eg, bone disease, infections, malignancies, cardiovascular complications) remained low, and patients had normal adult height, body mass index (BMI), and kidney function. Predictors of adult relapse included the number of relapses during childhood and the use of immunosuppressant medications other than steroids (ie, cyclosporine, chlorambucil, cyclophosphamide). [62]
Steroid-resistant nephrotic syndrome
Approximately 10% of patients overall with INS do not respond to an initial trial of steroids (2% of patients with MCNS do not respond to steroids). Additionally, about 1-3% of patients who initially do respond to steroids later become resistant to treatment ("late non-responders"). [2]
Most patients who do not achieve remission of proteinuria with steroids have kidney biopsy findings other than MCNS. The most common histopatholgical finding in these patients is FSGS. It is important to recognize that FSGS is not the name of a discrete disease entity but represents a pattern of injury. In fact, for a podocyte, it is the "end stage" wherein damaged podocytes detach from the glomerular basement membrane and the denuded membrane collapses or becomes adherent to the parietal epithelia of the Bowman's capsule. The segmental lesion undergoes sclerosis. Since podocytes are terminally differentiated, once detached, they are not replaced by a stem cell.
More than 60% of patients with nephrotic syndrome and FSGS who fail to achieve remission with any treatment progress to end-stage kidney disease (ESKD). In contrast, only 15% of patients with FSGS who achieve remission by any treatment progress to ESKD. [63] Gipson et al reported a 90% lower risk of progression to ESKD in patients with INS who achieved remission. [64]
Thus, patients with steroid-resistant INS have a good prognosis if remission of proteinuria can be achieved by medications other than corticosteroids. Failure to respond to treatment (ie, failure to achieve remission) and kidney insufficiency at presentation are predictors of poor outcome and progression to ESKD. [65]
General complications
Complications of INS include the following:
-
Edema
-
Hyperlipidemia
-
Thrombosis (renal vein thrombosis, deep vein thrombosis, and pulmonary embolism are the most frequently encountered thromboembolic complications in children; other venous sites of thrombosis include the superior sagittal sinus, other cerebral venous sites, and the inferior vena cava)
-
Infection (spontaneous bacterial peritonitis, sepsis, cellulitis)
-
Acute kidney injury
-
Adverse effects of medications (steroids, diuretics, albumin, steroid-sparing agents)
Patient Education
Soon after nephrotic syndrome is diagnosed, the patient and the family should be educated about the disease, its management, and its expected course. The family should participate in therapeutic decisions and should be encouraged to adhere to the medical regimen.
As with all chronic illnesses, many psychosocial issues may need to be addressed, including (but not limited to) the following:
-
Behavior
-
Adherence to medication
-
Adequate parental/caretaker supervision
-
Medical insurance
-
Missed work and school due to hospitalizations and outpatient visits
Consultation with social workers and mental health care workers may be useful.
Links to resources for parents can be found at the Web sites for the American Society of Pediatric Nephrology (ASPN) and the National Kidney Foundation.
-
Schematic drawing of the glomerular barrier. Podo = podocytes; GBM = glomerular basement membrane; Endo = fenestrated endothelial cells; ESL = endothelial cell surface layer (often referred to as the glycocalyx). Primary urine is formed through the filtration of plasma fluid across the glomerular barrier (arrows); in humans, the glomerular filtration rate (GFR) is 125 mL/min. The plasma flow rate (Qp) is close to 700 mL/min, with the filtration fraction being 20%. The concentration of albumin in serum is 40 g/L, while the estimated concentration of albumin in primary urine is 4 mg/L, or 0.1% of its concentration in plasma. Courtesy of the American Physiological Society (www.the-aps.org) [Haraldsson B, Nystrom J, Deen WM. Properties of the glomerular barrier and mechanisms of proteinuria. Physiol Rev. 2008 Apr;88(2):451-87.]
-
Mutational screening in children with isolated steroid-resistant nephrotic syndrome (SRNS). If next-generation sequencing (NGS) technology is accessible, screening should utilize a gene panel including, but not limited to, the most common monogenic causes of SRNS. If NGS technology is not available, genes should be screened in numerical order of frequency per age group. Ethnicity and histologic findings should trigger preferential screening of certain genes. DMS = diffuse mesangial sclerosis. Courtesy of Pediatric Nephrology (Open Access journal) [Preston R, et al. Genetic testing in steroid-resistant nephrotic syndrome: why, who, when and how? Pediatr Nephrol. 2019 Feb;34(2):195-210.]
Tables

What would you like to print?
- Overview
- Presentation
- DDx
- Workup
- Treatment
- Approach Considerations
- Corticosteroid Therapy
- Diuretic Therapy
- Antihypertensive Therapy
- Home Monitoring
- Frequently Relapsing and Steroid-Dependent Disease
- Steroid-Resistant Disease and Focal Segmental GS
- Investigational Treatments
- Side Effects of Drug Therapy
- Indications for Hospital Admission
- Diet and Activity
- Vaccination
- Consultations and Long-Term Monitoring
- Show All
- Guidelines
- Medication
- Questions & Answers
- Media Gallery
- References