Practice Essentials
Multicystic dysplastic kidney (MCDK), a variant of renal dysplasia, is one of the most frequently identified congenital anomalies of the urinary tract. Renal ultrasonography is the recommended initial diagnostic imaging study. The role of nephrectomy in the treatment of multicystic dysplastic kidney is controversial. [1]
Multicystic dysplastic kidney is the most common cause of an abdominal mass in the newborn period and is the most common cystic malformation of the kidney in infancy. See the image below.
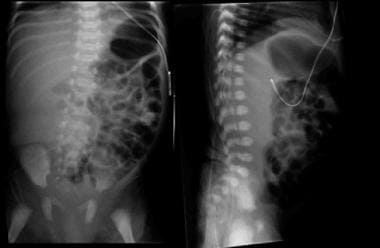 Kidney, ureter, and bladder (KUB) images of an infant with a right multicystic dysplastic kidney demonstrate displacement of bowel loops away from the right abdomen.
Kidney, ureter, and bladder (KUB) images of an infant with a right multicystic dysplastic kidney demonstrate displacement of bowel loops away from the right abdomen.
Signs and symptoms
Multicystic dysplastic kidney is usually asymptomatic and can remain undetected into adulthood. Abdominal or flank pain and respiratory distress are uncommon symptoms because of the pressure effect of the abnormal kidney.
Before the advent of fetal ultrasonography, an abdominal mass in the flank of an otherwise healthy newborn was the most common clinical presentation of unilateral multicystic dysplastic kidney.
Urinary malformations associated with multicystic dysplastic kidney include the following:
-
Bladder wall diverticulum
-
Contralateral renal agenesis
-
Dysplasia
-
Hypoplasia
-
Crossed fused renal ectopia
-
Cystic dysplasia of the testis
-
Ectopic kidney
-
Fibromuscular dysplasia
-
Horseshoe kidney
-
Patent urachus
-
Seminal vesicle abnormalities
-
Ureteropelvic junction obstruction
-
Urethral valves
See Presentation for more detail.
Diagnosis
Laboratory studies
A urine sample for dipstick and microscopic analysis should be obtained in patients with suspected multicystic dysplastic kidney. Urine should be sent for culture as needed.
Blood tests for creatinine, urea, and electrolytes should be performed.
Imaging studies
The following imaging studies may be included in the workup:
-
Renal ultrasonography
-
Voiding cystourethrography
-
Dimercaptosuccinic acid renal scanning
-
Technetium Tc 99m diethylenetriamine pentaacetic acid or technetium Tc 99m mercaptoacetyltriglycine renal scanning
Most cases of multicystic dysplasia of the kidney are detected during fetal ultrasonography and are reported as early as 15 weeks' gestation.
See Workup for more detail.
Management
The traditional reasons to consider nephrectomy for multicystic dysplastic kidney are to treat or prevent abdominal or flank pain, urinary tract infection, hypertension, or renal malignancy. The main controversy regarding the indications for nephrectomy is whether this procedure prevents renal malignancy.
See Treatment for more detail.
Background
Renal dysplasia, defined as abnormal metanephric differentiation, has variable presentations that cover a spectrum of conditions, including hypoplasia, multicystic dysplasia, and aplasia. Overall, renal dysplasia is the leading cause of end-stage renal disease in children.
Multicystic dysplastic kidney is a form of renal dysplasia characterized by the presence of multiple, noncommunicating cysts of varying size separated by dysplastic parenchyma and the absence of a normal pelvocaliceal system. The condition is associated with ureteral or ureteropelvic atresia, and the affected kidney is nonfunctional. Other terms used to describe this condition include multicystic kidney and multicystic renal dysplasia.
Pathophysiology
A brief overview of nephrogenesis is provided to highlight the developmental defects that lead to multicystic dysplastic kidney.
The urogenital system is predominantly derived from the intermediate mesoderm of the early embryo. [2] The intermediate mesoderm undergoes epithelial transformation to form the nephric (Wolffian) duct, which extends rostrocaudally in the embryo, adjacent to a tract of mesoderm (the nephrogenic cord). The nephric duct induces the adjacent nephrogenic cord mesenchyme to aggregate and transform into epithelial tubules.
During its caudal migration, the nephric duct induces 3 embryonic kidneys in the nephrogenic cord: pronephros, mesonephros, and metanephros (in a spacial and temporal sequence). When the nephric duct reaches the level of the developing hind limb, it gives rise to a caudal diverticulum, the ureteric bud that invades the metanephric mesenchyme in week 4 of human development.
Metanephros, which becomes the ultimate kidney is the product of reciprocal interactions between the metanephric mesenchyme and the ureteric bud. The metanephric mesenchyme forms the proximal components of the nephron from the glomerulus to the distal convoluted tubule. The ureteric bud that invades and branches inside the mesenchyme forms the distal components of the nephron, including the collecting ducts, calyces, pelvis, and ureter.
According to the leading hypothesis, the ureteric bud theory proposed by Mackie and Stephens, multicystic dysplastic kidney results from an abnormal induction of the metanephric mesenchyme by the ureteral bud. [3] This abnormal induction might be due to a problem with the formation of the mesonephric duct, the malformation of the ureteric bud, or the degeneration of the ureteric bud at an early stage. The final structure of the dysplastic kidney depends on the timing of the injury to the ureteric bud and on the effect of the injury on the ureteric bud branching.
Multicystic dysplastic kidney usually develops as a sporadic problem, although familial occurrence has been reported. [4] Mutations in genes important in ureteric bud development have been identified in syndromes with renal dysplasia, including multicystic dysplastic kidney. Specifically, mutations in EYA1 or SIX1 genes that lead to branchio-oto-renal (BOR) syndrome are associated with renal malformations, including multicystic dysplastic kidney. [5, 6] Mutations in the PAX2 gene, the cause of renal-coloboma syndrome (RCS), are associated with renal dysplasia. Hereditary multicystic dysplastic kidney was found in 3 generations of a family that also carried a PAX2 gene mutation. [7]
PAX2 mutations have also been identified in patients with isolated renal hypoplasia/dysplasia. Using predictive analysis, a study identified 2 new sequence variations: a deletion causing a frameshift (c.69delC) and a nucleotide substitution determining a splice site mutation (c.410+5 G/A). These findings suggest that all patients with kidney and urinary tract malformations may benefit from PAX2 molecular analysis. [8]
Exposure to viral infections in utero has been associated with multicystic dysplastic kidney. Cytomegalovirus (CMV), enterovirus, and adenovirus have been implicated in the development of renal dysplasia. Teratogens may also play a role in abnormal renal development, although their association with multicystic dysplastic kidney has not been clearly established.
Urinary tract obstruction during fetal development causes urinary stasis, cyst formation, and disruption of nephrogenesis; however, the degree of obstruction does not correlate with the severity of dysplasia. Thus, obstruction is not proven as a mechanism of multicystic dysplastic kidney.
Multicystic dysplastic kidney may persist without any change, may increase in size, or may undergo spontaneous involution. Calcification may develop in persistent multicystic dysplastic kidney, particularly in adults, and has been reported as early as age 3 months. Most cases of unilateral multicystic dysplastic kidney undergo spontaneous involution. The change may be due to resorption of the fluid within the cysts.
Multicystic dysplastic kidney can be diagnosed prenatally. [9, 10, 11] An involution of prenatally diagnosed multicystic dysplastic kidney has been noted before birth. Some cases of prenatally diagnosed multicystic dysplastic kidney monitored prior to birth demonstrate an initial increase in size followed by involution.
The Multicystic Kidney Registry reported 260 patients with multicystic dysplastic kidney whose cases were managed nonoperatively and whose cases were followed for varying periods as long as 5 years. [12] Approximately 18% of these kidneys were undetectable by age 1 year, 31% were undetectable by age 3 years, and 54% were undetectable by age 5 years. The initial ultrasonographic evidence of involution is a change in cyst distension that precedes a decrease in renal size.
Whether involution of the cysts is associated with involution of the intervening renal parenchyma is unclear. The residual parenchyma may be too small to be identified with conventional diagnostic imaging studies. Avni et al reported a child with prenatally diagnosed multicystic dysplastic kidney who had an operation when aged 3 months without repeat ultrasonography. [13] At surgery, neither renal tissue nor renal vasculature was found.
Etiology
Multicystic dysplastic kidney has been reported in various syndromes, including the following:
-
49,XXXXX syndrome: Patients with this syndrome present with hypertelorism, epicanthic folds, microcephaly, short neck, clinodactyly of the fifth finger, small hands and feet, and intellectual disability.
-
Alagille syndrome: Alagille syndrome is an autosomal dominant condition characterized by liver disease, cardiac defects, and characteristic facies, including a prominent forehead and pointed chin. Involvement of one or more additional systems, such as kidney, eye, skeleton, vasculature, and pancreas, is common. Renal anomalies include cystic dysplasia. The condition is associated with mutations in the JAG1 gene that encodes for a ligand of Notch.
-
Beckwith-Wiedemann syndrome: Patients with this syndrome present with macrosomia, microcephaly, macroglossia, visceromegaly, omphalocele, and hypoglycemia.
-
Branchio-oto-renal (BOR) syndrome: The BOR syndrome is an autosomal dominant condition caused by mutations in the EYA1 and SIX1 genes; patients usually present with hearing loss, ear malformations (eg, preauricular pits), branchial cleft fistulas or cysts, and dysplasia or renal agenesis. Renal dysplasia is more common than renal agenesis.
-
Hypoparathyroidism-deafness-renal syndrome: This autosomal dominant syndrome is caused by mutations in the GATA3 transcription factor. Patients present with hypoparathyroidism, sensorineural deafness, and urinary tract anomalies, including renal dysplasia.
-
Joubert syndrome: This syndrome is an autosomal recessive condition characterized by hypoplasia of the cerebellar vermis, hypotonia, and impaired psychomotor development together with abnormal respiratory pattern, abnormal eye movements with poor vision, or both.
-
Maturity-onset diabetes of the young type V (MODY5): MODY5 is a monogenic form of diabetes with an autosomal dominant mode of inheritance caused by mutations in the hepatocyte nuclear factor 1-beta mutations. Patients present with diabetes, usually when younger than 25 years, with a wide spectrum of renal anomalies, including cystic dysplasia.
-
Renal coloboma syndrome (RCS): The RCS is an autosomal dominant condition caused by mutations in the transcription factor PAX2 and characterized by optic nerve coloboma and renal malformations, including dysplasia.
-
Trisomy 18: Patients with trisomy 18 have a prominent occiput, micrognathia, low-set ears, flexion deformities of the fingers, congenital heart disease, and intellectual disability.
-
VACTERL association: The VACTERL association refers to the combination of vertebral defects (V), anal atresia (A), cardiovascular anomalies (C), tracheoesophageal fistula (TE), renal anomalies (R), and limb defects (L). In a study of 50 patients with the VACTERL association, 11 (22%) had cystic renal disease. [14]
-
Waardenburg syndrome type 1: Patients present with developmental anomalies of the eyelids, eyebrows, and nose root; pigmentary defects of the iris and hair; and congenital deafness.
-
Williams syndrome: This syndrome is characterized by elfin facies, intellectual disability, supravalvular aortic stenosis, and neonatal hypercalcemia.
Gastrointestinal malformations reported in patients with multicystic dysplastic kidney include the following:
-
Inguinal hernia
-
Tracheoesophageal fistula
Neurologic malformations reported in patients with multicystic dysplastic kidney include the following:
-
Caudal agenesis
-
Caudal regression syndrome
-
Congenital deafness
-
Intellectual disability
-
Microphthalmia
-
Myelomeningocele
Cardiovascular malformations reported in patients with multicystic dysplastic kidney include the following:
-
Pulmonary stenosis
-
Truncus arteriosus
Musculoskeletal malformations reported in patients with multicystic dysplastic kidney include the following:
-
Clinodactyly of the fifth finger
-
Congenital dislocation of the hip
-
Flexion deformities of the fingers
-
Talipes equinovarus
Miscellaneous malformations reported in patients with multicystic dysplastic kidney include the following:
-
Bipartite uterus
-
Epicanthic folds
-
Hymenal atresia
-
Hypertelorism
-
Low-set ears
-
Macroglossia
-
Micrognathia
-
Pigmentary defects of iris and hair
-
Preauricular pit
-
Short neck
Urinary malformations associated with multicystic dysplastic kidney include the following:
-
Bladder wall diverticulum
-
Contralateral renal agenesis
-
Dysplasia
-
Hypoplasia
-
Crossed fused renal ectopia
-
Cystic dysplasia of the testis
-
Ectopic kidney
-
Fibromuscular dysplasia
-
Horseshoe kidney
-
Patent urachus
-
Seminal vesicle abnormalities
-
Ureteropelvic junction obstruction (UPJO): Contralateral UPJO has been reported in 7-12% of patients.
-
Urethral valves
-
Vesicoureteral reflux (VUR): Contralateral VUR is reported in 4-19% of patients. [15, 16] Ipsilateral VUR might also be present. [17] VUR is usually low grade and typically resolves in early life.
Atiyeh et al retrospectively reviewed 56 patients with multicystic dysplastic kidney and noted associated urinary abnormalities in 29 patients (52%). [18]
Several authors have suggested that multicystic dysplastic kidney might be subcategorized based on the presence or absence of associated contralateral urinary abnormalities. De Klerk et al suggested that multicystic dysplastic kidney associated with atresia of the urinary pelvis is not associated with significant contralateral urinary abnormalities, whereas multicystic dysplastic kidney associated with atresia of a lower ureteral segment is associated with significant contralateral urinary abnormalities and has a poorer prognosis. [19]
Bloom et al suggested that the incidence of contralateral urinary abnormalities might vary with the size of the multicystic dysplastic kidney–affected kidney. [20] These authors reported that a large kidney with multicystic dysplastic kidney is not associated with contralateral urinary abnormalities, whereas a small kidney with multicystic dysplastic kidney is associated with contralateral urinary abnormalities and with other congenital anomalies.
The contralateral kidney is usually larger than normal. In children older than 2 years, 72% of patients with multicystic dysplastic kidney showed compensatory growth of the contralateral kidney. Compensatory growth of the contralateral kidney has been noted in utero and was noted in 8 (24%) of 33 children at birth. [21]
Epidemiology
International statistics
The incidence of unilateral multicystic dysplastic kidney is reported to be 1 in 4300 live births, and the combined incidence of unilateral and bilateral multicystic dysplastic kidney is 1 in 3600 live births. Up to 39% of patients present with associated anomalies in the contralateral kidney. [22] Bilateral multicystic dysplastic kidney occurs in about 20% of prenatally diagnosed cases of multicystic dysplastic kidney. The left kidney is involved in 55% of cases, and the right kidney is involved in 45%.
Sex-related demographics
A review of 14 studies that reported on 340 patients with unilateral multicystic dysplastic kidney revealed a male-to-female ratio of 1.48:1. [23]
Prognosis
Prognosis depends on whether the involvement is unilateral or bilateral and on the presence and severity of associated anomalies. Most individuals with isolated unilateral multicystic dysplastic kidney do not experience any problems or complications as a consequence of this congenital abnormality.
Morbidity/mortality
Multicystic dysplastic kidney may persist without any change, may increase in size, or may undergo spontaneous involution. Calcification may develop in persistent multicystic dysplastic kidney, particularly in adults, and has been reported as early as age 3 months. Most cases of unilateral multicystic dysplastic kidney undergo spontaneous involution. Morbidity and mortality are uncommon in patients with a unilateral multicystic dysplastic kidney and a normal contralateral kidney. Morbidity and mortality may result from urinary tract infection (UTI), hypertension, or neoplasia.
Complications
The following complications of multicystic dysplastic kidney might be categorized as those due to multicystic dysplastic kidney and those due to associated urinary malformations:
-
Abdominal or flank pain
-
Urinary tract infection (UTI)
-
Hypertension
-
Renal malignancy
-
Gastric outlet obstruction and respiratory depression
Urinary tract infection
The Multicystic Kidney Registry reported UTIs in 12 (5%) of 260 patients who were observed for 5 years. [12] Pyelonephritis, when present, is almost always on the contralateral side and is often associated with VUR.
UTI in patients with multicystic dysplastic kidney is rare because the ureteral atresia presumably prevents ascending infection; however, Hartman et al reported abscess formation in multicystic dysplastic kidney, and Reitelman et al reported an individual with a primarily infected kidney with multicystic dysplastic kidney. [24, 25] The latter authors presumed an ascending infection developed when bacteria traversed the microlumen of an atretic ureteric segment.
Hypertension
The Multicystic Kidney Registry reported mild hypertension in 4 (1.5%) of 260 individuals with multicystic dysplastic kidney who were observed for 5 years. [12]
A systematic review of 29 studies reported 6 cases of hypertension in 1115 eligible children with multicystic dysplastic kidney. The mean probability of a child with unilateral multicystic dysplastic kidney developing hypertension was 5.4 cases per 1000. [26] Although the risk of hypertension in multicystic dysplastic kidney is low, the results of this study did not allow firm recommendations on the frequency and duration of blood pressure measurement follow-up for children with multicystic dysplastic kidney. Thus, prospective cohort study with a long follow-up is needed.
Seeman et al performed ambulatory blood pressure monitoring on 25 children with multicystic dysplastic kidney. [27] Five (20%) children had blood pressures greater than the 95th percentile, 2 had combined daytime and nighttime hypertension, and 3 had isolated nighttime hypertension. Hypertension was more common in children with contralateral urinary abnormalities.
Ambrose et al identified 4 adults with hypertension that did not improve following nephrectomy of the kidney with multicystic dysplastic kidney. [28]
Hypertension that resolved after nephrectomy of an multicystic dysplastic kidney–affected kidney has been reported in 4 individuals. In one case, the plasma renin activity was elevated before nephrectomy and normalized after nephrectomy. [29]
Most patients with multicystic dysplastic kidney have no radiologically demonstrable blood flow. A nonfunctional bloodless kidney is an unlikely cause of hypertension in infancy and early childhood. However, the presence of a contralateral congenital urinary abnormality such as ureteropelvic junction obstruction (UPJO) or renal dysplasia, the development of a pyelonephritic scar in a contralateral kidney with VUR, or the effects over time of hyperfiltration of the contralateral kidney are potential causes of hypertension.
Renal malignancy
Nephroblastoma (Wilms tumor) in multicystic dysplastic kidney has been reported in 7 children with an average age of 7 months.
Renal cell carcinoma was reported in 13 patients at an average age of 39 years, and an embryonal tumor was reported in a 68-year-old patient. [22]
Nodular renal blastema is reported in 0.25-0.5% of the general population, 3-6.7% of individuals with multicystic dysplastic kidney, and 12-40% of patients with Wilms tumor. [30] The increased incidence of nodular renal blastema in multicystic dysplastic kidney might confer a higher risk for Wilms tumor.
The intervening stroma of a kidney affected by multicystic dysplastic kidney may not undergo complete involution and may provide a focus for malignant degeneration. However, no increased risk of Wilms tumor has been noted in multicystic dysplastic kidney. A systemic review of 26 studies demonstrated no cases of Wilms tumor in 1041 children with unilateral multicystic dysplastic kidney. [31] These data are consistent with an earlier study by the American Multicystic Kidney Disease Registry that found no cases of renal neoplasia in 260 patients with multicystic dysplastic kidney. [32]
Beckwith noted that nodular renal blastema is present in as much as 1% of the general population and that Wilms tumor develops in approximately 1 in 8000 children. [30] Approximately 1 in 80 infants with nodular renal blastema develops Wilms tumor. The incidence of nodular renal blastema in multicystic dysplastic kidney is considered to be approximately 5%; therefore, 20 multicystic dysplastic kidney–affected kidneys would need to be removed to ablate one with nodular renal blastema and 1600 kidneys with multicystic dysplastic kidney would need to be removed to prevent one case of Wilms tumor. Because the cure rate for Wilms tumor is approximately 90%, 16,000 multicystic dysplastic kidney–affected kidneys would need to be removed to save one life. Based on this analysis, Beckwith does not recommend nephrectomy to prevent the development of Wilms tumor in a patient with multicystic dysplastic kidney.
In 2015, the Canadian Urological Society updated their guidelines following a 2014 literature review to obtain up-to-date information regarding the medical implications of having a multicystic dysplastic kidney, including complications and appropriate follow-up to reduce the risk of complications. [33]
Gastric outlet obstruction and respiratory depression
Gastric outlet obstruction with feeding difficulties and respiratory depression due to elevation of the hemidiaphragm are rare reported complications.
Patient Education
All patients with multicystic dysplastic kidney should be counseled on the lifetime implications of the presence of only one functional kidney. Women with fetal multicystic dysplastic kidney may benefit from genetic counseling, supported by findings that a substantial proportion of patients with multicystic dysplastic kidneys have abnormal microarray analysis. [34, 35]
-
Kidney, ureter, and bladder (KUB) images of an infant with a right multicystic dysplastic kidney demonstrate displacement of bowel loops away from the right abdomen.






