Background
Morquio syndrome (mucopolysaccharidosis type IV [MPS IV]) is a rare lysosomal storage disease (LSD) that is inherited in an autosomal-recessive fashion. Morquio syndrome is classified into 2 types, Morquio A syndrome (mucopolysaccharidosis type IVA [MPS IVA]; OMIM 253000) and Morquio B syndrome (mucopolysaccharidosis type IVB [MPS IVB]; OMIM 253010), based on a deficiency of different lysosomal enzymes—N-acetylgalactosamine-6-sulfate sulfatase (GALNS) and β-galactosidase, respectively. GALNS deficiency induces the accumulation of glycosaminoglycans (GAGs), keratan sulfate (KS) and chondroitin-6-sulfate (C6S) in multiple tissues, particularly bone, cartilage, heart valves, and cornea, whereas β-galactosidase deficiency induces the accumulation of only KS in those tissues. [1, 2, 3, 4, 5, 6]
Morquio syndrome is characterized by a unique skeletal dysplasia with excessive KS and/or C6S accumulation. Although most individuals with Morquio syndrome appear normal at birth, skeletal abnormalities often develop within the first year of life. At birth, some affected newborns may have a minor skeletal phenotype, such as humpback, chest protrusion, and prominent forehead, as confirmed with radiography. [3, 4, 5, 6, 7]
Skeletal dysplasia is progressive and characterized by incomplete ossification and successive imbalance of growth, which results in a prominent forehead, abnormal face with a large mandible, short neck, cervical spine instability, gibbus thoracolumbar spinal deformity, tracheal deviation and obstruction, disproportionate short-trunk dwarfism, pectus carinatum, flaring of the rib cage, hip dysplasia with coxa valga, genu valgum, hypermobile joints, and pes planus. [3, 4, 5, 6, 8, 9, 10, 11] The severity of bone and cartilage damage differs by the severity of the phenotype (mild, intermediate, or severe) and the type of bone affected. Endochondral bone formation seems to be affected in MPS IV.
In 1929, Luis Morquio first reported 4 Swedish patients with MPS IV (now classified as MPS IVA). [3] In the same year, Brailsford also reported a patient with MPS IV. [4] Clinical features of this patient included prominent forehead, abnormal face with a large mandible, short neck, pectus carinatum, flaring of the rib cage, hypermobile joints, genu valgum, disproportionate short-trunk dwarfism, and pes planus; however, aortic valve disease and corneal clouding were not described in the original publication. [3]
In 1962, Pedrini et al isolated and identified KS in the urine of 3 patients with Morquio syndrome and reported that this metabolic disorder differs from that observed in Hurler syndrome (mucopolysaccharidosis type I [MPS I]). [5] In 1965, McKusick et al classified Hurler and Hunter syndromes, as well as Morquio syndrome, as hereditary acid mucopolysaccharidoses (MPS I to MPS V). MPS V was referred to as Scheie Syndrome. [12]
In 1971, Orii et al reported an attenuated (intermediate) form of MPS IVA. [6] At 5 months old, adduction of both thumbs was recognized, and at age 18 months, a chest abnormality was noticed. At age 3, the patient had kyphosis, and an abnormal gait was present by age 5. The patient had keratosulfaturia and was diagnosed enzymatically with Morquio A syndrome at age 15. At age 18, he was 135 cm tall, had corneal clouding, [1] and exhibited milder skeletal deformities, such as pectus carinatum, hypermobile joints, and genu valgum.
In 1974, the GALNS enzyme and its deficiency were discovered and identified using oligosaccharide substrate prepared from C6S containing N-acetylgalactosamine-6-sulfate. [11] In 1976, O’Brien et al reported a patient with a mild clinical status similar to Morquio A syndrome. However, this patient was deficient in β-galactosidase. [13] In 1977, Arbisser et al reported a similar case with mild Morquio A syndrome–like symptoms resulting from a deficiency of β-galactosidase, described as MPS IVB. [14] Morquio syndrome has been differentiated into Morquio A syndrome and Morquio B syndrome.
In 1981, Orii et al reported a very mild form of Morquio A syndrome in 2 siblings. [15] Their initial symptom was hip joint pain at age 8; thus, both patients underwent femoral osteotomy at age 13. Both patients had keratosulfaturia, mild thorax changes, and corneal clouding, although they did not show unique pectus carinatum, genu valgum, excessive joint laxity, or facial changes. Initial radiographic studies in both cases revealed mild platyspondyly, slight anterior wedging of the lumbar vertebra, minimal odontoid hypoplasia, and subtle capital femoral epiphysis. However, when they were aged 18 and 22, the ossified femoral heads disappeared with erosion and widening of the femoral necks. These signs were more significant in the older brother. The 2 brothers were not diagnosed with Morquio A syndrome until ages 29 and 25; their heights were 147 and 157 cm, respectively. In 2015, they were 52 and 57 years, respectively, and their clinical condition was stable. GALNS enzyme activity in the fibroblasts of these patients was about 10% of that seen in cells from healthy controls. [16]
Morquio A syndrome is a heterogeneous disease. Patients exhibit mild to severe phenotypes with different degrees of systemic bone dysplasia (see image below). [6, 8, 9, 10, 15, 16, 17, 18, 19, 20, 21, 22]
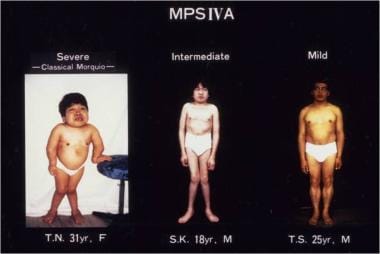 Clinical pictures of patients with Morquio A syndrome (mucopolysaccharidosis type IVA [MPS IVA]). Left panel: a 31-year-old female patient with a severe form. Middle panel: an 18-year-old male patient with an intermediate form. Right panel: a 25-year-old male patient with a mild form. Image adapted originally from Educational CD for the International Morquio Organization; contributed by Shunji Tomatsu, MD, PhD, and reviewed by Mary C Theroux, MD, and William G Stuart Mackenzie, MD, MA, Nemours Children's Health.
Clinical pictures of patients with Morquio A syndrome (mucopolysaccharidosis type IVA [MPS IVA]). Left panel: a 31-year-old female patient with a severe form. Middle panel: an 18-year-old male patient with an intermediate form. Right panel: a 25-year-old male patient with a mild form. Image adapted originally from Educational CD for the International Morquio Organization; contributed by Shunji Tomatsu, MD, PhD, and reviewed by Mary C Theroux, MD, and William G Stuart Mackenzie, MD, MA, Nemours Children's Health.
Individuals with untreated severe Morquio A syndrome die of respiratory obstruction, cervical spinal cord compression, or heart valve disease in their second or third decade of life. The lifespan of individuals with the attenuated form of Morquio A syndrome is as long as 70 years. [10, 17] The height prognosis is poor among individuals with severe bone dysplasia. Intellectual impairment does not occur in Morquio A syndrome.
Historically, Morquio B syndrome was considered to have milder manifestations than Morquio A syndrome. The clinical manifestations of Morquio B syndrome include platyspondyly, femoral epiphyses, atlantooccipital instability, genu valgum, gait abnormalities, and corneal clouding. [1] Morquio B syndrome is associated with normal or near-normal stature, normal neck development, absence of hearing loss, and hepatomegaly. [13, 14] Some cases of Morquio B have also presented with neuronopathic manifestations such as cognitive delay and dystonia. [23] The skeletal involvement in Morquio B syndrome is less pronounced than in Morquio A syndrome. However, radiographic and other phenotypical analyses cannot differentiate between Morquio A and Morquio B syndrome because of the extensive overlap, which depends on the skeletal symptoms, the enzyme activity, recognition of complications, and response to surgery, among other factors.
The genetic mutations of the GALNS gene leading to the prevalence of Morquio A are diverse. More than 400 mutations have been discovered associated with the development of this syndrome, and patients with Morquio A are found globally. [24, 25, 26] Studies have indicated a founder effect within particular populations worldwide, such as the British/Irish, Japanese, and Colombian populations. [27, 28, 29] Additionally, a founder effect was reported in Morquio B Spanish population. [27]
Pathophysiology
Mucopolysaccharidoses (MPS) are a group of lysosomal storage diseases caused by deficiencies of lysosomal enzymes. Excessive accumulation of GAGs such as dermatan sulfate (DS), heparan sulfate (HS), chondroitin sulfate (CS), and KS in multiple tissues leads to coarse facial features, CNS damage, organomegaly, connective tissue disorders, and skeletal dysplasia. GAGs accumulate in lysosomes, intracellular matrix, and extracellular matrix. MPS is categorized into 8 groups, with 12 different deficient lysosomal enzymes. [30]
In Morquio syndrome, the degradation of KS is defective because of a deficiency of either GALNS in Morquio A syndrome or β-galactosidase in Morquio B syndrome (see image below). Deficiency of GALNS also impairs the catabolism of C6S. KS and C6S have various physiological and biological functions. Recent studies have determined that untreated MPS patients demonstrate an increase in IL-6 and TNF-α, indicating an inflammatory response. [23, 31] Morquio A and Morquio B syndromes are inherited through an autosomal-recessive trait. KS is closely involved in cellular motility, embryo implantation, wound healing, corneal transparency, and nerve regeneration. [21] C6S is also involved in embryo development, maintaining mechanical skin strength and cell proliferation (see image below).
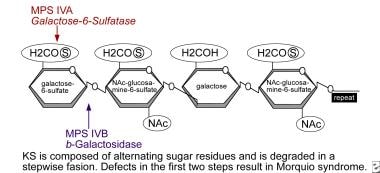 Defects in keratan sulfate (KS) degradation resulting in Morquio syndrome. Stepwise degradation of KS; each type or subtype corresponds to a deficiency in a different enzyme involved in the degradation route. *Galactose-6-sulfatase (namely, N-acetylgalactosamine-6-sulfate sulfatase; GALNS): The sulfatase that cleaves the sulfate from galactose residues is deficient in patients with MPS IVA. Image adapted originally from Educational CD for the International Morquio Organization; contributed by Shunji Tomatsu, MD, PhD, and reviewed by Mary C Theroux, MD, and William G Stuart Mackenzie, MD, MA, Nemours Children's Health.
Defects in keratan sulfate (KS) degradation resulting in Morquio syndrome. Stepwise degradation of KS; each type or subtype corresponds to a deficiency in a different enzyme involved in the degradation route. *Galactose-6-sulfatase (namely, N-acetylgalactosamine-6-sulfate sulfatase; GALNS): The sulfatase that cleaves the sulfate from galactose residues is deficient in patients with MPS IVA. Image adapted originally from Educational CD for the International Morquio Organization; contributed by Shunji Tomatsu, MD, PhD, and reviewed by Mary C Theroux, MD, and William G Stuart Mackenzie, MD, MA, Nemours Children's Health.
Epidemiology
Morquio A Syndrome
The incidence of Morquio A syndrome has been reported as 1 per 216,000 births in British Columbia, [32] 1 per 76,000 births in Northern Ireland, [33] 1 per 201,000 births in Australia, [34] 1 per 450,000 births in Portugal, [35] 1 per 640,000 births in Western Australia, [36] 0.15 per 100,000 live births in Brazil, [37] 1.10 per 100,000 live births in Mexico, [38] and 1 per 625,000 births in Japan. [10] Incidence in the United States has been estimated at 0.11 in 100,000 live births. [39] Morquio A is the third most common MPS in India, the United States, and most of Europe; and the second most common MPS behind MPS II in Southern and Eastern Europe. [40]
Morquio B Syndrome
The incidence of Morquio B syndrome is much lower than that of Morquio A syndrome. Based on the information provided by the National MPS Society, only 3 patients were registered between 1995–2015 in the United States. [39] In Brazil, the incidence has been estimated as 0.003 per 100,000 live births. [37]
Prognosis
Patients with the severe form of Morquio syndrome can develop cervical myelopathy early and do not survive past their second or third decade of life if untreated. [17, 41, 42] A shorter-than-usual lifespan may be attributed to paralysis caused by myelopathy, restrictive chest movement, and valvular heart disease. [41, 43] Two thirds of patients die of respiratory distress, whereas the second leading cause of death in Morquio A disease is cardiac failure. [43, 44] Cardiovascular manifestations in Morquio A disease are evidenced by recent findings on alterations in vessel compliance and severe vascular intima-media thickening. [45]
Patients with mild manifestations of Morquio syndrome (MPS IV), regardless of type, have been reported to survive into the seventh decade of life.
The lifespan of patients with Morquio A syndrome is improving due to advancements in comprehensive care. Additionally, enzyme replacement therapy (ERT) and hematopoietic stem cell transplantation (HSCT) for Morquio A disease address some systemic manifestations, in some cases slowing the progression of the disease [46] and improving the overall quality of life, especially if treatment starts at an early age. [47, 48, 49, 50, 51]
Patient Education
Many resources are available to patients with Morquio syndrome and their families to increase their understanding of the disorder, including the following:
-
Little People of America (LPA)
-
Morquio conference hhps:// morquioconference.wixsite.com/morquio/
It is important to educate the patient’s primary physicians, teachers, counselors, school administrators, and classmates to help them understand the unique needs of an individual with Morquio syndrome.
Genetic Counselors should get involved in the educational process and provide tailored education to the patients and their families (https://www.nsgc.org/). Morquio syndrome does not affect fertility, and a child born to a parent with Morquio syndrome will be a carrier but will not have Morquio syndrome unless he or she partners with another Morquio syndrome carrier. Then, the risk is 25% that their offspring will have Morquio syndrome. As in all autosomal-recessive conditions, if both parents have the same subtype of Morquio syndrome, they will have a 100% chance of having offspring with Morquio syndrome.
-
Clinical pictures of patients with Morquio A syndrome (mucopolysaccharidosis type IVA [MPS IVA]). Left panel: a 31-year-old female patient with a severe form. Middle panel: an 18-year-old male patient with an intermediate form. Right panel: a 25-year-old male patient with a mild form. Image adapted originally from Educational CD for the International Morquio Organization; contributed by Shunji Tomatsu, MD, PhD, and reviewed by Mary C Theroux, MD, and William G Stuart Mackenzie, MD, MA, Nemours Children's Health.
-
Defects in keratan sulfate (KS) degradation resulting in Morquio syndrome. Stepwise degradation of KS; each type or subtype corresponds to a deficiency in a different enzyme involved in the degradation route. Numbered reactions correspond to specific disorders. *Galactose-6-sulfatase (namely, N-acetylgalactosamine-6-sulfate sulfatase; GALNS): The sulfatase that cleaves the sulfate from galactose residues is deficient in patients with MPS IVA. Image adapted originally from Educational CD for the International Morquio Organization; contributed by Shunji Tomatsu, MD, PhD, and reviewed by Mary C Theroux, MD, and William G Stuart Mackenzie, MD, MA, Nemours Children's Health.
-
Clinical features of a patient with Morquio A syndrome (MPS IVA). This patient had a severe form of MPS IVA at age 3 years and had bone abnormalities of pectus carinatum, kyphoscoliosis, genu valgum, short stature, prominent forehead, and abnormal gait (height, 90 cm; 50th percentile of male MPS IVA growth chart; body weight, 14 kg). Image adapted originally from Educational CD for the International Morquio Organization; contributed by Shunji Tomatsu, MD, PhD, and reviewed by Mary C Theroux, MD, and William G Stuart Mackenzie, MD, MA, Nemours Children's Health.
-
Cervical spine, flexion (A) and extension (B) views, in a child aged 5 years 11 months. These flexion and extension images depict anterior and posterior subluxation, respectively, of the atlas secondary to odontoid hypoplasia.
-
Bilateral lower extremity views in a patient aged 22 years 6 months. Metaphyseal irregularities and the characteristic genu valgus deformity are easily observed in this image.
-
MRI of the cervical spine in a 4-year-old patient. A baseline study of the upper cervical anatomy is recommended no later than 2 years or at diagnosis using flexion/extension radiography. If radiographs demonstrate atlantoaxial (C1-2) instability, or if the child has concern for neck pain, weakness in the extremities, or an abnormal neurologic exam, an MRI of the cervical spine in both neck flexion and extension is recommended to assess for spinal cord compression. Regular follow-up and annual radiographs with MRI, as needed, are required. Image adapted originally from Educational CD for the International Morquio Organization; contributed by Shunji Tomatsu, MD, PhD, and reviewed by Mary C Theroux, MD, and William G Stuart Mackenzie, MD, MA, Nemours Children's Health.
-
Clinical picture of hip deformity in an 8-year-old patient with Morquio A syndrome (MPS IVA). Multiple abnormalities are shown in the hip, including proximal femoral epiphyseal dysplasia, dysplastic (shallow) acetabula with coxa valgus deformity, and flared iliac wings. Image adapted originally from Educational CD for the International Morquio Organization; contributed by Shunji Tomatsu, MD, PhD, and reviewed by Mary C Theroux, MD, and William G Stuart Mackenzie, MD, MA, Nemours Children's Health.
-
Skeletal/joint disease - hands. Bilateral hand radiographs in a 6-year-old patient with MPS IVA. Note the tapering of the proximal portion of metacarpals 2 through 5 and small irregular carpal bones. The joints may become hyperlax by age 2 years. Eventually, the hands take on a characteristic tilting of the radial epiphysis toward the ulna due to a combination of metaphyseal deformities, hypoplasia of the bones, and degradation of connective tissues near the joint secondary to GAG accumulation. Image adapted originally from Educational CD for the International Morquio Organization; contributed by Shunji Tomatsu, MD, PhD, and reviewed by Mary C Theroux, MD, and William G Stuart Mackenzie, MD, MA, Nemours Children's Health.
-
Skeletal/joint disease - hands. The tapered irregular distal radius and ulna exemplify the epiphyseal involvement characteristic of Morquio syndrome. Overall, the bones are osteopenic with cortical thinning. Upper extremities in a child aged 2 years and 3 months (left panel). Note the irregular epiphyses and widened metaphysis. Cortical thinning and mild widening of the diaphysis of the humerus are visible. With aging, the bone deformity progresses, eg, with the tilting of the radial epiphysis toward the ulna. The humerus usually appears shortened later. Image adapted originally from Educational CD for the International Morquio Organization; contributed by Shunji Tomatsu, MD, PhD, and reviewed by Mary C Theroux, MD, and William G Stuart Mackenzie, MD, MA, Nemours Children's Health.
-
Skeletal/joint disease - hands. Unlike other mucopolysaccharidoses, Morquio syndrome is associated with ligamentous laxity. The cause of abnormal joint function remains unknown, presumably derived from a combination of metaphyseal deformities, hypoplasia of the bones, and degradation of connective tissues near the joint secondary to GAG accumulation. The wrist and fingers (small joints) are usually fragile, resulting in a weak grip. Difficulties with dressing, personal hygiene, and writing can result from this hypermobile ligament. Range-of-motion exercises, swimming, and computer typing offer some benefits in preserving joint function and fine motor skills and should be started early in the clinical course. Wrist splints and plastic braces may benefit fine motor functioning. The indication of physical therapy and its benefits in Morquio A syndrome should be studied further. Image adapted originally from Educational CD for the International Morquio Organization; contributed by Shunji Tomatsu, MD, PhD, and reviewed by Mary C Theroux, MD, and William G Stuart Mackenzie, MD, MA, Nemours Children's Health.
-
Skeletal/joint problem – hands and legs. This 10-year-old patient with Morquio A syndrome (MPS IVA) showed skeletal habitus with a disproportionate short stature of short trunk type with relatively long extremities. The upper extremities show lateral bowing and prominence of the radius. In the lower extremities, there is genu valgum (knock knee). A skew foot posture is seen in both feet, as well as an increased sandal gap between toes 1 and 2. Extreme ligamentous laxity permits very hypermobile knees, fingers, and wrists. The feet show pronated pes planus. Muscle bulk is reduced, particularly in the extremities. Generalized mild to moderate hypotonia is present. Image adapted originally from Educational CD for the International Morquio Organization; contributed by Shunji Tomatsu, MD, PhD, and reviewed by Mary C Theroux, MD, and William G Stuart Mackenzie, MD, MA, Nemours Children's Health.
-
Spinal cord pathophysiology. Odontoid hypoplasia is the most critical skeletal feature to be recognized in any patient with Morquio syndrome (MPS IV). This, in combination with ligamentous laxity and extradural mucopolysaccharide deposition, results in atlantoaxial subluxation, with consequential quadriparesis or even death. Another potential complication is cervical myelopathy. A history of exercise intolerance in patients with MPS IV often predicts occult cervical myelopathy, which can cause bowel and bladder dysfunction and spinal cord compression, leading to weakness or paralysis. Mortality and morbidity are primarily related to atlantoaxial instability and subsequent cervical myelopathy, and patients with a severe form, primarily related to cervical instability, often do not survive beyond the second or third decade of life. A minor fall or neck extension can result in spinal cord injury and subsequent quadriparesis or sudden death. Image adapted originally from Educational CD for the International Morquio Organization; contributed by Shunji Tomatsu, MD, PhD, and reviewed by Mary C Theroux, MD, and William G Stuart Mackenzie, MD, MA, Nemours Children's Health.
-
Pathophysiology of difficult airway in Morquio A syndrome. Both restrictive and obstructive respiratory pathologies are common in patients with Morquio A syndrome. The restrictive defect results from thoracic cage deformity, and the obstructive defect is caused by tracheobronchial abnormalities, large tongue and mandible, adenoidal, tonsillar, and vocal cord hypertrophy by the accumulation of storage materials. An imbalance of growth between trachea, cervical spine, and brachiocephalic artery causes tracheal obstruction. Moreover, individuals with Morquio A syndrome have small nasal passages caused by thickened mucous membranes and thick and copious secretions. Chronic upper respiratory tract infection further decreases the already diminished airway lumen. Image adapted originally from Educational CD for the International Morquio Organization; contributed by Shunji Tomatsu, MD, PhD, and reviewed by Mary C Theroux, MD, and William G Stuart Mackenzie, MD, MA, Nemours Children's Health.
-
Tracheal obstruction. CT angiography in a 29-year-old patient shows severe tracheal obstruction. Tracheal narrowing (T; trachea) increases with age. Note the position of the brachiocephalic artery (A) anterior to the trachea, which is commonly observed. However, the brachiocephalic artery only partially contributes to the narrowing of the trachea. In some cases, there is no such contribution by the Brachiocephalic artery, and the narrowing in those cases is entirely due to other factors. The cervicothoracic spine moves forward while a severe pectus carinatum (M: manubrium) compresses backward. Image adapted originally from Educational CD for the International Morquio Organization; contributed by Shunji Tomatsu, MD, PhD, and reviewed by Mary C Theroux, MD, and William G Stuart Mackenzie, MD, MA, Nemours Children's Health.
-
Gene therapy approaches. Created with BioRender.com; contributed by Betul Celik, MS.
-
Flowchart to navigate the steps to reach an accurate diagnosis.
-
Patient with MPS IVA. Coronal and sagittal reconstructions from a computed tomography (CT) scan show hypoplasia of the odontoid process of C2. Image adapted originally from Educational CD for the International Morquio Organization; contributed by Shunji Tomatsu, MD, PhD, and reviewed by Mary C Theroux, MD, and William G Stuart Mackenzie, MD, MA, Nemours Children's Health.
-
Morquio A syndrome symptoms and possible misdiagnosis with the algorithm for definitive diagnosis on suspicion. Created with BioRender.com; contributed by Fnu Nidhi, MS.
-
GAGs in the trachea and humerus of a 23-year-old male patient with Morquio A. (A) and (B) Trachea tissue stained with AB-PAS, GAGs are shown in dark blue. (C) and (D) The right humerus with colloidal iron, and GAGs are also shown in blue. Courtesy of ScienceDirect/Elsevier [Doherty C, Averill LW, Theroux M, et al. Natural history of Morquio A patient with tracheal obstruction from birth to death. Molecular Genetics and Metabolism Reports. March 2018;14:59-67. Online at https://www.sciencedirect.com/journal/molecular-genetics-and-metabolism-reports/vol/14/suppl/C.]
-
Sections of the weight-bearing area of femoral heads from a 17-year-old female Morquio A patient. (A) H&E staining. (B) Safranin O Staining (C) Masson staining. Courtesy of Frontiers Media [Ma Y, Peng H, Hsiang F, et al. Case Report: Diagnosis of Mucopolysaccharidosis Type IVA With Compound Heterozygous Galactosamine-6 Sulfatase Variants and Biopsy of Replaced Femoral Heads. Front Pediatr. 4 July 2022;10:914889. Online at https://www.frontiersin.org/articles/10.3389/fped.2022.914889/full.]
-
Lateral view of spine of a child aged 8 years 7 months with Morquio A syndrome. This radiograph shows advanced platyspondyly, irregularity, and anterior beaking of vertebral bodies characteristic of dysostosis multiplex. Image adapted originally from Educational CD for the International Morquio Organization; contributed by Shunji Tomatsu, MD, PhD, and reviewed by Mary C Theroux, MD, and William G Stuart Mackenzie, MD, MA, Nemours Children's Health.
-
A 34-year-old patient uses two styles of Med-El audio processors (Sonnet and Rondo); the patient is wearing her cochlear implant processor SONNET (left panel) and RONDO (right panel). Rondo is an all-in-one unit design that uses disposable batteries for up to 75 h. The patient usually uses Rondo when she travels because of its long battery life. She uses a behind-the-ear design SONNET more regularly than RONDO since batteries are rechargeable (despite shorter battery life of 6 or 10 h), and it is challenging to lose.
-
Dental Problems. A 24-year-old patient with MPS IVA has widely spaced and spade-shaped incisiors. Attention to daily oral hygiene and professional dental cleaning and evaluation every 6-12 months is necessary to minimize the effects of thin dental enamel. Image adapted originally from Educational CD for the International Morquio Organization; contributed by Shunji Tomatsu, MD, PhD, and reviewed by Mary C Theroux, MD, and William G Stuart Mackenzie, MD, MA, Nemours Children's Health.
-
Accumulation of the storage material in foam cell, chondrocyte, and endothelial cell (EM). (A) Foam cell in bone marrow (arrow) (6,000x). (B) Vacuolated chondrocyte (arrow) in humerus (7,500x). ECM is thick and disorganized. (C) Endothelial cell with vacuoles (arrow) in heart (15,000x). (D) Vacuolated chondrocyte (arrow) in trachea (4,000x). ECM is thick and disorganized.
-
Appearance of vacuolated cells in femoral cap from a 17-year-old female patient with Morquio B (stained toluidine blue with 0.5 µm section; light microscopy). Her height was 127 cm and her body weight was 34 kg. She had compound heterozygous mutations with p.R482H (c.1445 G>A/G) and p.T500A (c.1498 A>A/G). (A) Vacuolated cells and chondrocytes in bone marrow; arrows show the vacuolated cells. (B) Vacuolated osteocytes (arrows).
-
Systems biology. Contributed by Amali Karunathilaka, BS.
-
Defects in keratan sulfate (KS) degradation resulting in Morquio syndrome. Stepwise degradation of KS; each type or subtype corresponds to a deficiency in a different enzyme involved in the degradation route. *Galactose-6-sulfatase (namely, N-acetylgalactosamine-6-sulfate sulfatase; GALNS): The sulfatase that cleaves the sulfate from galactose residues is deficient in patients with MPS IVA. Image adapted originally from Educational CD for the International Morquio Organization; contributed by Shunji Tomatsu, MD, PhD, and reviewed by Mary C Theroux, MD, and William G Stuart Mackenzie, MD, MA, Nemours Children's Health.






