Practice Essentials
Perhaps the best way to consider mixed connective tissue disease is as an undifferentiated connective tissue disease represented mostly by Raynaud phenomenon and anti-RNP antibody. See the image below.
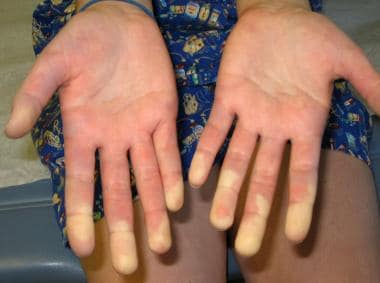 Raynaud phenomenon showing demarcation of color difference.
Raynaud phenomenon showing demarcation of color difference.
This disorder may evolve into one of several major connective tissue diseases or to an overlap syndrome of the major connective tissue diseases. The evolution of this disease requires the physician to carefully assess and constantly reassess the patient in anticipation of change and to provide early intervention with appropriate medical therapy.
Signs and symptoms
The most frequent presentation of mixed connective tissue disease is a child with polyarthritis, general malaise, and Raynaud phenomenon.
Patients may present with the following:
-
Sclerodermatous skin (usually limited to fingers but can be more extensive)
-
Sausage-shaped fingers
-
Proximal muscle weakness
-
Rash (finger ulcers or pits, Gottren papules)
-
Vasculitic rashes (usually palpable purpuric rashes)
-
Dysphagia
-
Gastroesophageal reflux disease (GERD) symptoms
-
Fever
-
Rheumatoid nodules
-
Lymphadenopathy
-
Alopecia
-
Telangiectasia
-
Headache
See Presentation for more detail.
Diagnosis
Laboratory studies
Diagnostic laboratory studies include the following:
-
Antinuclear antibody
-
Anti–double-stranded DNA
-
Autoantibody panel, including antibodies against ribonucleoprotein (RNP), Smith, Ro(SSA), La(SSB), Scl-70, phospholipids, cardiolipin and histone, total hemolytic complement, C3, C4, quantitative immunoglobulins, and thyroid studies
Imaging studies
Initial imaging studies should include the following:
-
Chest radiography
-
Barium swallow
-
Echocardiography
Other tests
Other tests include the following:
-
Baseline pulmonary function tests, including diffusing capacity of lung for carbon monoxide
-
Nailfold capillaroscopy, particularly in patients who have more scleroderma features
See Workup for more detail.
Management
The most important tools in the treatment of pediatric mixed connective tissue disease include tailoring the medical regimen, promptly attending to disease flares, and performing careful and frequent clinical and laboratory evaluations to test for new disease manifestations.
See Treatment and Medication for more detail.
Background
Sharp et al first proposed mixed connective tissue disease (MCTD) as a separate autoimmune disorder. [1] The initial definition identified patients with a specific autoantibody profile, high titers of anti-U1 ribonucleoprotein (70-kD) autoantibody (anti-RNP Ab) but without anti-Smith autoantibody (anti-Sm Ab), in association with specific clinical criteria. Alarcon-Segovia and Villareal and Kasukawa et al subsequently suggested 2 alternate sets of criteria. [2, 3] A comparison of the sensitivity and specificity of the above 3 sets of criteria along with a fourth set of criteria developed by Kahn et al demonstrated that the Kahn and Alarcon-Segovia criteria are the most sensitive and specific for disease diagnosis. [4]
In pediatrics, Kasukawa criteria are used most frequently in published series and have more conservative requirements. Mixed connective tissue disease remains a controversial diagnosis. Some rheumatologists view mixed connective tissue disease as a separate disease; others classify the disorder as an undifferentiated connective tissue disease or overlap syndrome, which may have features of lupus, progressive systemic sclerosis, rheumatoid arthritis, and myositis but should not have its own separate name. [5, 6]
Adding support to the concept of mixed connective tissue disease as a distinct entity, in 1993, Mairesse et al described an autoantibody to the constitutive 73-kD heat shock protein found at high levels exclusively in patients with mixed connective tissue disease. [4] This autoantibody was found in reduced levels in patients with progressive systemic sclerosis and rheumatoid arthritis. The autoantibody was not found in significant quantities in patients with systemic lupus erythematosus (SLE) or myositis. This finding has not been duplicated and must be interpreted with caution. However, the authors redefine mixed connective tissue disease as "a core of minor symptoms (ie, Raynaud phenomenon, puffy fingers, mild myositis, and arthritis) associated significantly with anti-U1-68kD antibody, defining an undifferentiated connective tissue (UCTD) disease that may ultimately overlap with features of major connective tissue disease."
Although confusing, perhaps the best way to consider mixed connective tissue disease is as an undifferentiated connective tissue disease represented mostly by Raynaud phenomenon and anti-RNP antibody.
This disorder may evolve into one of several major connective tissue diseases or to an overlap syndrome of the major connective tissue diseases. The evolution of this disease requires the physician to carefully assess and constantly reassess the patient in anticipation of change and to provide early intervention with appropriate medical therapy.
Mixed connective tissue disease has features of several autoimmune diseases. [7] For more details see Juvenile Rheumatoid Arthritis, Neonatal Lupus and Cutaneous Lupus Erythematosus in Children, Systemic Lupus Erythematosus, Systemic Sclerosis, Sjogren Syndrome, Dermatomyositis, and Myositis Ossificans.
Etiology
Specific causes of mixed connective tissue disease remain undefined. [8] Research suggests that many factors, including genetics, hormones, and environment, contribute to development of autoimmune syndromes.
The hallmark of mixed connective tissue disease is the presence of autoantibodies to U1 small nuclear ribonucleoproteins, in particular a 70-kDa U1 protein. During cell death or apoptosis, the 70-kDa protein is cleaved by caspase-3 into a small 40-kDa protein. Several research groups have described this apoptotic form.
Specific anti-RNP autoantibodies that preferentially bind to the apoptotic form of the U1 protein have been demonstrated in a group of patients with mixed connective tissue disease (29 of 53 tested). Furthermore, the concentration of the autoantibody is high early in the disease and decreases over time, suggesting that it correlates or represents an inciting event in disease onset.
Epidemiology
United States data
In a literature review, Michels counted 224 cases of mixed connective tissue disease. [9] Pediatric-onset mixed connective tissue disease accounts for an estimated one quarter of all cases. Most large pediatric rheumatology centers in major cities have 5-15 active pediatric cases, although some studies estimate that mixed connective tissue disease occurs in 0.6% of all pediatric rheumatology patients.
US data are derived from international data.
Race-, sex-, and age-related demographics
Ethnic distribution for pediatric mixed connective tissue disease has not been reported. Literature suggests that no specific protection or propensity based on race is noted.
A female predominance, which is typical of other autoimmune diseases, is noted in mixed connective tissue disease. Three published series on pediatric mixed connective tissue disease report 89 of 105 patients to be female, or a female-to-male ratio of approximately 6:1.
Age range for pediatric-onset disease is younger than 16 years by definition. No specific age of onset is excluded. The median age at onset is 12 years, based on reported pediatric series. The youngest reported age at onset is 2 years. A recent 15-year retrospective study concluded the mean age at disease onset was 10.7 years. [10]
Prognosis
Prognosis is generally considered similar to that of pediatric lupus. Initial descriptions of mixed connective tissue disease did not include renal disease, and the prognosis was believed to be considerably better than for the major connective tissue diseases. However, patients who fit the criteria for mixed connective tissue disease have had renal disease and considerable morbidity and mortality from major organ manifestations. It appears that, in mixed connective tissue disease, children fare better than adults.
Individual patients appear to have severe or mild disease courses.
Prognosis also depends on which disease manifestations are more prominent (eg, myocarditis, pulmonary disease, renal disease).
Morbidity/mortality
Literature describes pediatric mixed connective tissue disease from individual case reports to small series. Mortality is 0-50%. The review by Michels found a mortality figure of 7.6%. [9] More recent data assess pediatric mortality at 3-4 per 1000 population versus adult mortality at 12-23 per 1000 population. Serious organ involvement included 47% of patients with renal disease, 54% with restrictive lung disease, and 29% with GI disease. Although rare, morbidity from cerebral disease, cardiomyopathy, myopericarditis, and pulmonary hypertension has been reported and is associated with a significant risk of mortality.
Complications
Complications of mixed connective tissue disease depend on the organ systems involved and the adverse effects and risks of immunosuppressive therapy. Patients with mixed connective tissue disease are at risk for infections, cardiovascular disease, and complications observed in lupus, progressive systemic sclerosis, and myositis.
Witczak et al studied cardiac function in 50 patients with pediatric mixed connective tissue disease of 15 years’ duration. Both left and right ventricular dysfunction were detected. Left ventricular dysfunction was significantly associated with higher disease activity and longer prednisolone therapy. [11]
Patient Education
Patient and family must have a thorough understanding of the disease, potential severity, and complications from the disease and therapy. Treatment of the individual with mixed connective tissue disease is difficult, especially for adolescent patients. The physician, parents, and/or caregivers should expect issues including depression and noncompliance. They must be prepared to work together with the patient toward a better outcome.
Knight et al aimed to characterize the prevalence of depression and anxiety in pediatric systemic lupus erythematosus and mixed connective tissue disease patients and determine their association with healthcare utilization. Depression and anxiety symptoms were prevalent in patients with SLE/MCTD, and suicidal ideation significantly more common in SLE/MCTD than in healthy subjects. Despite prevalent symptoms, there were poor rates of prior mental health treatment, and less frequent PCP visits among those with depression symptoms. The authors concluded that further investigation of barriers to mental health care and interventional strategies for symptomatic youth with SLE/MCTD is needed. [12]
-
Raynaud phenomenon showing demarcation of color difference.








