Practice Essentials
Methemoglobinemia occurs when red blood cells (RBCs) contain methemoglobin at levels higher than 1%. Methemoglobin results from the presence of iron in the oxidized ferric form (Fe3+) instead of the usual reduced ferrous form (Fe2+). This results in a decreased availability of oxygen to the tissues. This condition can be congenital or acquired. [1]
Symptoms are proportional to the methemoglobin level. At levels up to 20%, changes can occur in the color of blood (see the image below) and skin. As levels rise above 20%, neurologic and cardiac symptoms arise as a consequence of hypoxia. Levels higher than 70% are usually fatal.
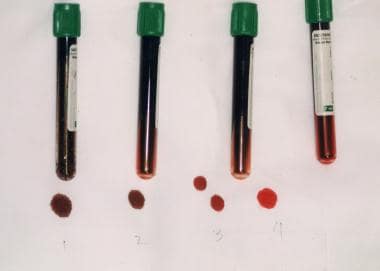 Note chocolate brown color of methemoglobinemia. In tubes 1 and 2, methemoglobin fraction is 70%; in tube 3, 20%; and in tube 4, normal.
Note chocolate brown color of methemoglobinemia. In tubes 1 and 2, methemoglobin fraction is 70%; in tube 3, 20%; and in tube 4, normal.
Key elements of the history include the following:
-
In congenital methemoglobinemia, characteristic diffuse persistent slate-gray cyanosis, often present from birth; patients are often asymptomatic despite the presence of cyanosis
-
In acute methemoglobinemia, which is usually acquired, a history of exposure to methemoglobinemia-inducing substances such as certain prescription drugs, local anesthetics, and occupational exposure; however, this information is not always available
-
Any known family history of methemoglobinemia or glucose-6-phosphate dehydrogenase (G6PD) deficiency
Symptoms are proportional to the fraction of methemoglobin and the resulting hypoxia. A normal methemoglobin fraction is about 1% (range, 0-3%). Symptoms associated with higher levels of methemoglobin are as follows:
-
< 10% - None (patients with underlying diseases may have more symptoms at lower level)
-
10-20% - Slight discoloration (eg, pale, gray, blue) of the skin
-
20-30% - Anxiety, headache, tachycardia, lightheadedness
-
30-50% - Dyspnea, weakness, confusion, chest pain
-
50-70% - Arrhythmias; altered mental status, delirium, seizures, coma; profound acidosis
-
> 70% - Usually, death [2]
Physical findings may include the following:
-
Discoloration of the skin and blood (the most striking physical finding)
-
Cyanosis – This occurs in the presence of 1.5 g/dL (10%) of methemoglobin (as compared with 5 g/dL of deoxygenated hemoglobin)
-
Seizures
-
Coma
-
Dysrhythmia (eg, bradyarrhythmia or ventricular dysrhythmia)
-
Acidosis
-
Cardiac or neurologic ischemia
-
Pallor of the skin or conjunctiva (suggestive of anemia and possible hemolysis)
-
Skeletal abnormalities and intellectual disability
See Presentation for more detail.
Diagnosis
Laboratory studies that may be ordered include the following:
-
Studies to rule out hemolysis – Complete blood count (CBC), reticulocyte counts, lactate dehydrogenase (LDH), indirect bilirubin, haptoglobin
-
Studies to test for organ failure and general end-organ dysfunction – Liver function tests, electrolyte concentrations, blood urea nitrogen (BUN), creatinine
-
Urine pregnancy tests
-
Heinz body preparation (indicative of oxidative injury to the erythrocyte)
-
Hemoglobin electrophoresis to identify hemoglobin M (Hb M); some difficult cases require DNA sequencing of the globin chain gene or mass spectrometry for diagnosis
-
Specific enzyme assays for causative deficiencies
-
Bedside tests for methemoglobinemia - Examination of blood color on white filter paper after exposure to room air or after aerating a tube of blood with 100% oxygen; if the blood remains dark with these maneuvers, then methemoglobinemia is likely
-
Serum levels of nitrites or other offending drugs
Oxygen-carrying capacity of the blood may be determined with the help of the following:
-
Arterial blood gas (ABG) determination
-
Pulse oximetry (SpO 2): Typically less accurate than CO-oximetry in the setting of methemoglobinemia, with the exception of newer multiwavelength pulse oximeters
-
CO-oximetry (if available): Measures concentration of oxyhemoglobin, carboxyhemoglobin, methemoglobin, and reduced hemoglobin
There may be a discrepancy between the SpO2 and the oxygen concentration calculated in arterial blood gas (SaO2). If the methemoglobin level is above 10-15%, SpO2 will remain depressed, at approximately 85% even if the patient is on 75-90% oxygen; however, SaO2 will be falsely nomal, as this technology considers all hemoglobin to be oxyhemoglobin or deoxyhemoglobin. [3]
Other studies that may be considered are as follows:
-
Potassium cyanide test to distinguish methemoglobin from sulfhemoglobin
-
CT of the head
-
Chest radiography to exclude pulmonary or cardiac disease
-
Echocardiography to determine the presence of congenital heart disease
See Workup for more detail.
Management
Early clinical recognition of methemoglobinemia is essential. Treatment is determined by the symptoms:
-
Severe methemoglobinemia can be life-threatening and necessitate emergency therapy
-
Chronic mild methemoglobinemia may be completely asymptomatic and necessitate no specific therapy
-
No pharmacologic treatment exists for hereditary forms of methemoglobinemia
Initial care includes the following:
-
Administration of supplemental oxygen
-
Determination of the underlying etiology (eg, toxin or drug)
-
Removal of the offending oxidizing substance
Treatment is advisable for patients who have suffered acute exposure to an oxidizing agent and have methemoglobin levels of 20% or higher, as well as for those with lower methemoglobin levels who have significant comorbidities (eg, cardiac, pulmonary, or hematologic disease), especially in the presence of end-organ dysfunction.
Treatment modalities include the following:
-
Methylene blue – The primary emergency treatment for documented symptomatic methemoglobinemia (contraindicated in G6PD deficiency and ineffective with hemoglobin M)
-
Exchange transfusion - Can be considered for patients who do not respond to methylene blue or G6PD-deficient individuals who are severely symptomatic
-
Hyperbaric oxygen treatment – Another option when methylene blue therapy is ineffective or contraindicated
-
IV hydration and bicarbonate (for metabolic acidosis)
-
Other medications – These include ascorbic acid, riboflavin, and cimetidine
-
Dietary measures – Avoidance of precipitants in food or drink
See Treatment and Medication for more detail.
Background
Methemoglobin contains iron in the ferric state (Fe3+) rather than the reduced ferrous form (Fe2+) found in hemoglobin. This structural change causes an alteration in the blood’s ability to bind oxygen. Methemoglobin is a naturally occurring oxidized metabolite of hemoglobin, and physiologic levels (< 1%) are normal. Problems arise as methemoglobin levels increase. Methemoglobin does not bind oxygen, thus effectively leading to a functional anemia. [4, 5, 6]
In addition, methemoglobin causes a leftward shift of the oxygen-hemoglobin dissociation curve, resulting in decreased release of oxygen to the tissues. The presence of anemia and cyanosis despite oxygen treatment results from both of these effects. [7] (See Pathophysiology and Etiology.)
Methemoglobinemia occurs when red blood cells (RBCs) contain methemoglobin at levels higher than 1%. This may be from congenital causes, increased synthesis, or decreased clearance. Increased levels may also result from exposure to toxins that acutely affect oxidation-reduction reactions, resulting in increasing methemoglobin levels.
Clinically, methemoglobinemia has a variable course (see Presentation). Because of the nonspecificity of the clinical findings, mild cases may go undiagnosed. Fatigue, flulike symptoms, and headaches may be the only manifestations in the initial phase. Symptoms are proportional to the methemoglobin level and include skin color changes (cyanosis with blue or grayish pigmentation) and blood color changes (brown or chocolate color). As levels of methemoglobin rise above 15%, neurologic and cardiac symptoms arise as a consequence of hypoxia. Levels higher than 70% are usually fatal. [2]
Tests to rule out hemolysis and to test for organ failure and general end-organ dysfunction should be performed. Urine pregnancy tests should be performed in females of childbearing age. Tests to evaluate a hereditary cause for methemoglobinemia should be ordered when appropriate. (See Workup.)
The most important aspects of the management of methemoglobinemia are recognition of the condition and prompt initiation of treatment, when indicated. For mild asymptomatic cases, treatment is purely for cosmetic or psychological reasons. When methemoglobinemia is severe or symptomatic, specific therapy may be indicated. Initial care includes supplemental oxygen and removal of the offending oxidizing substance. Various agents can reduce the methemoglobin levels to within the reference range, or at least to acceptable levels. (See Treatment.)
Pathophysiology
RBCs contain hemoglobin, which has a quaternary structure. Each hemoglobin molecule is composed of 4 polypeptide chains. Each of these chains is associated with a heme group, which contains iron in the reduced or ferrous form (Fe2+). In this form, iron can combine with oxygen by sharing an electron, thus forming oxyhemoglobin. When oxyhemoglobin releases oxygen to the tissues, the iron molecule is restored to its original ferrous state.
Hemoglobin can accept and transport oxygen only when the iron atom is in its ferrous form. When hemoglobin loses an electron and becomes oxidized, the iron atom is converted to the ferric state (Fe3+), resulting in the formation of methemoglobin. Methemoglobin lacks the electron that is needed to form a bond with oxygen and thus is incapable of oxygen transport.
Under normal conditions, methemoglobin levels remain below 1%; however, under conditions that cause oxidative stress, levels will rise. The low level of methemoglobin is maintained through 2 important mechanisms. The first is the hexose-monophosphate shunt pathway within the erythrocyte. Through this pathway, oxidizing agents are reduced by glutathione.
The second and more important mechanism involves two enzyme systems, diaphorase I and diaphorase II, and requires nicotinamide adenine dinucleotide (NADH) and nicotinamide adenine dinucleotide phosphate (NADPH), respectively, to reduce methemoglobin to its original ferrous state.
NADH-dependent methemoglobin reduction (diaphorase I pathway) is the major enzymatic system involved. [8] Cytochrome b5 reductase plays a major role in this process by transferring electrons from NADH to methemoglobin, an action that results in the reduction of methemoglobin to hemoglobin. This enzyme system is responsible for the removal of 95-99% of the methemoglobin that is produced under normal circumstances.
NADPH-dependent methemoglobin reduction (diaphorase II pathway) usually plays only a minor role in the removal of methemoglobin. This enzyme system utilizes glutathione production and glucose-6-phosphate dehydrogenase (G6PD) to reduce methemoglobin to hemoglobin. It assumes a larger and more important role in methemoglobin regulation in patients with cytochrome b5 reductase deficiencies.
The NADPH-dependent methemoglobin reduction pathway can be accelerated by exogenous cofactors such as methylene blue to as much as five times its normal level of activity. [6, 9, 8, 10] In the absence of further accumulation of methemoglobin, these methemoglobin reduction pathways can clear methemoglobin at a rate of approximately 15% per hour.
Acquired methemoglobinemia is considerably more common than congenital forms.
Congenital (hereditary) methemoglobinemia
At least two forms of congenital cytochrome b5 reductase deficiency exist. Both are inherited in an autosomal recessive pattern. In type I b5R deficiency, the more common form, cytochrome b5 reductase is absent only in RBCs. Homozygotes appear cyanotic but usually are otherwise asymptomatic. Methemoglobin levels typically range from 10% to 35%. Life expectancy is not adversely influenced, and pregnancies are not complicated. Heterozygotes may develop acute, symptomatic methemoglobinemia after exposure to certain drugs or toxins.
The type II b5R form is substantially less common, accounting for only 10-15% of cases of congenital cytochrome b5 reductase deficiency. In this condition, cytochrome b5 reductase is deficient in all cells, not just RBCs. It is associated with several other medical problems, including intellectual disability, microcephaly, and other neurologic complications. Life expectancy is severely compromised, and patients usually die at a very young age. The exact mechanism of the neurologic complications is not known.
Methemoglobinemia may also involve the presence of abnormal hemoglobins (hemoglobin M [Hb M]). In most of these hemoglobins, tyrosine replaces the histidine residue, which binds heme to globin. This replacement displaces the heme moiety and permits oxidation of the iron to the ferric state. Consequently, Hb M is more resistant to reduction by the methemoglobin reduction enzymes (see above). This results in a functionally impaired hemoglobin with a decreased affinity for oxygen.
The inheritance pattern for Hb M variants is autosomal dominant, whereas that for methemoglobinemia due to cytochrome b5 reductase deficiency is autosomal recessive. Patients with Hb M appear cyanotic but are otherwise generally asymptomatic. There are three phenotypic varieties of Hb M, corresponding to the globulin gene affected (alpha, beta, or gamma) as follows [8, 11, 12] :
-
Alpha-chain variants cause neonatal cyanosis that is persistent
-
Beta-chain variants do not cause cyanosis until several months after birth, when the level of fetal hemoglobin has decreased
-
Gamma-chain variants cause transient neonatal cyanosis that resolves once the level of fetal hemoglobin decreases
Another hemoglobin variant, hemoglobin E (Hb E), is associated with methemoglobinemia as well. In a study from the National Thalassemia Center in Sri Lanka, 45 patients who had a diagnosis of Hb E beta-thalassemia were found to have significantly higher median methemoglobin levels that normal control subjects and patients with other hemoglobinopathies (2.7% vs 0.3%.) Furthermore, methemoglobin levels were significantly elevated in patients who had undergone a previous splenectomy. [13]
Acquired methemoglobinemia
Acquired methemoglobinemia is much more common than the congenital form and involves excessive production of methemoglobin. Often, it is associated with the use of or exposure to oxidant drugs, chemicals, or toxins, including dapsone, [14] local anesthetic agents, [15] and nitroglycerin. This increased production overwhelms the normal physiologic regulatory and excretory mechanisms. These oxidant agents can cause an increase in methemoglobin levels either by ingestion or by absorption through the skin. A study involving two tertiary care teaching hospitals indicated that methemoglobinemia was present in a significant proportion of all hospitalized patients, and the frequency may be much more than anticipated or expected. [16]
The presence of methemoglobin may also be a marker and predictor of sepsis, resulting from release of excessive amounts of nitrous oxide (NO). [17]
Clinical manifestations
Organs with high oxygen demands (eg, the central nervous system [CNS] and the cardiovascular system) usually are the first systems to manifest toxicity. Oxygenated blood is bright red, deoxygenated blood is dark red, and blood containing methemoglobin is dark reddish brown (see the image below). This dark hue is responsible for clinical cyanosis.
Note chocolate brown color of methemoglobinemia. In tubes 1 and 2, methemoglobin fraction is 70%; in tube 3, 20%; and in tube 4, normal.
Clinical evidence of cyanosis depends on the level of methemoglobin. Skin discoloration can occur in patients who are not anemic when as little as 1.5 g/dL (about 10%) of hemoglobin is in the form of methemoglobin. By comparison, a deoxyhemoglobin level of 5 g/dL is required to produce clinical cyanosis. When methemoglobin levels are relatively low, cyanosis may be observed without cardiopulmonary symptoms.
In methemoglobinemia, cyanosis is usually the first presenting symptom. In other conditions associated with cyanosis resulting from hypoxemia, it is a much later finding.
In patients with severe anemia, a higher percentage of methemoglobin is required for cyanosis to be obvious. These patients are more likely to exhibit signs of hypoxemia and have less cyanosis than is seen in patients who do not have anemia.
Etiology
Congenital (hereditary) methemoglobinemia
Hereditary methemoglobinemias may be divided into two categories, as follows [2] :
-
Methemoglobinemia due to an altered form of hemoglobin (ie, Hb M)
-
Methemoglobinemia due to an enzyme deficiency (NADH reductase deficiency) that decreases the rate of reduction of iron in the hemoglobin molecule
Several variants of hemoglobin M have been described, including Hb Ms, Hb MIwate, Hb MBoston, Hb MHyde Park, and Hb MSaskatoon. These are usually autosomal dominant in nature. Alpha-chain substitutions cause cyanosis at birth, whereas the effects of beta-chain substitutions become clinically apparent in infants at 4-6 months of age.
There are four types of hereditary methemoglobinemias that are secondary to deficiency of NADH cytochrome b5 reductase, which is encoded by the CYB5R3 gene. All of them are autosomal recessive disorders. Heterozygotes have 50% enzyme activity and no cyanosis; homozygotes who have elevated methemoglobin levels above 1.5% have clinical cyanosis. The four types are as follows:
-
Type I – This is the most common variant, and the enzyme deficiency is limited to the erythrocytes causing cyanosis; cyanosis usually, but not always, develops during infancy. [18]
-
Type II – Widespread deficiency of the enzyme occurs in various tissues, including erythrocytes, liver, fibroblasts, and brain; it is associated with severe CNS symptoms, including encephalopathy, microcephaly, hypertonia, athetosis, opisthotonos, strabismus, intellectual disability, and growth retardation; cyanosis is evident at an early age.
-
Type III – Although the entire hematopoietic system (platelets, RBCs, and white blood cells [WBCs]) is involved, the only clinical consequence is cyanosis.
-
Type IV – As in type I, involvement is limited to the erythrocytes. This type results in chronic cyanosis
Deficiency of NADPH-flavin reductase can also cause methemoglobinemia.
Acquired methemoglobinemia
Acquired methemoglobinemia is more common. It is usually due to the ingestion of drugs or toxic substances. Exposure to such substances in amounts that exceed the enzymatic reduction capacity of RBCs precipitates symptoms. [16] Acquired methemoglobinemia is more frequent in premature infants and infants younger than 4 months, and the following factors may have a role in the higher incidence in this age group:
-
Fetal hemoglobin may oxidize more easily than adult hemoglobin
-
The level of NADH reductase is low at birth and increases with age; it reaches adult levels by age 4 months
-
Higher gastric pH in infants may facilitate bacterial proliferation, resulting in increased conversion of dietary nitrates to nitrites
-
An association between methemoglobinemia and acute gastroenteritis in infants has been noted in several studies. This may be due to acidosis from loss of stool bicarbonate, which impairs the already immature function of the methemoglobin reductase system in these young patients
Benzocaine, phenazopyridine, dapsone, and nitrates/nitrites are the most common substances associated with methemoglobinemia. [19] Organic and inorganic nitrites can be absorbed through the skin, and many prescription cardiac medications contain these compounds. Treatment of preterm infants with inhaled nitric oxide may lead to methemoglobinemia; susceptibility to methemoglobinemia in these infants may be increased by carriage of a single-nucleotide polymorphism in the CYB5R3 gene. [20]
Dietary intake may occur in infants or adults who ingest well water that has been contaminated with nitrites caused by water runoff from fertilized fields. [21] Sodium nitrite is used as a food preservative, and prepackaged foods may contain significant levels of nitrites. [22, 23] Intentional ingestion of sodium nitrite has been increasingly reported as a method of suicide. [24, 25]
Methemoglobinemia has been reported in infants fed purées of vegetables (borage and chard) with high levels of nitrates. [26] Tests confirmed that vegetables with the highest nitrate levels Consumption of vegetables (eg, spinach, beets, carrots) that were contaminated with bacteria and inadequately cooked has been associated with methemoglobinemia. [27] Infants and patients on gastric acid−reduction therapy are particularly vulnerable to methemoglobinemia because gastric acid production may not be sufficient to maintain low levels of nitrate-reducing bacteria in the intestine.
Fava bean ingestion in patients with G6PD deficiency is another potential dietary cause of methemoglobinemia. [28] Favism has been reported in breastfed infants with G6PD deficiency whose mother consumed fava beans. [29]
Medarov et al reported methemoglobinemia associated with dialysis sessions using a portable dialysis unit in five critically ill hospitalized patients. The episodes were traced to inadequate clearance of the disinfectant chloramine from the tap water used for the dialysis. [30]
Chlorates are another group of oxidizing agents that can cause methemoglobinemia. These substances are found in matches, explosives, and fungicides.
Topical and injected local anesthetics (eg, benzocaine, [16, 31] lidocaine, [32] prilocaine, phenazopyridine, [33, 34] cerium nitrate, and silver sulfadiazine [35] ) have also caused methemoglobinemia. Predisposing factors for the development of this toxicity include the presence of a mucosal injury with resultant increased absorption or a previously undiagnosed methemoglobin reductase enzyme deficiency. This toxicity can also be idiosyncratic.
Acquired methemoglobinemia caused by use of cocaine laced with benzocaine has been reported. [36]
A 10-year retrospective case-control study by Chowdhary and colleagues of 94,694 procedures in which topical anesthetics were used identified 33 cases of methemoglobinemia, for an incidence of 0.035%. However, risk was increased in hospitalized patients and those who received benzocaine-based anesthetics. The procedures included bronchoscopy, nasogastric tube placement, esophagogastroduodenoscopy, transesophageal echocardiography, and endoscopic retrograde cholangiopancreatography. [37]
Dapsone, a drug used to prevent and treat Pneumocystis jirovecii pneumonia (PCP) and to treat leprosy and other skin diseases (including a topical preparation used for acne [38] ), has also been associated with methemoglobinemia. This drug should be used with great caution in patients with known G6PD deficiency, methemoglobin reductase deficiency, or hemoglobin M. [16, 39]
Rasburicase treatment for tumor lysis syndrome in patients with low catalase activity (inherited or acquired) may result in methemoglobinemia secondary to the formation of hydrogen peroxide. [40, 41, 42] Some authors have suggested measuring catalase activity before initiating rasburicase therapy in this setting.
RBCs in patients with liver cirrhosis undergo severe oxidative stress, especially in the setting of bleeding complications. [43] The level of methemoglobin is significantly higher in the RBCs of these patients than in those of nonbleeding patients.
Idiopathic methemoglobinemia can occur in association with systemic acidosis. This typically occurs in infants younger than 6 months and is usually caused by dehydration and diarrhea. Idiopathic methemoglobinemia is exacerbated by the lower levels of methemoglobin reductase enzyme found in infants (50% of adult levels).
A case study suggested methemoglobinemia as a potential adverse effect of the combination of pembrolizumab and axitinib for treatment of metastatic renal cell carcinoma (most likely that complication was related to axitinib). [44]
Other substances that can cause methemoglobinemia include the following:
-
Copper sulfate
-
Analgesics and antipyretics – Acetaminophen, [54] acetanilide, phenacetin, and celecoxib
-
Zopiclone [55]
-
Methylene blue (high dose or in G6PD-deficient patients [58] )
-
Indigo carmine
-
Resorcinol
-
Metoclopramide [59]
-
Antibiotics – Sulfonamides, nitrofurans, and para-aminosalicylic acid
-
Industrial/household agents – Aniline dyes, nitrobenzene, naphthalene (moth balls), aminophenol, and nitroethane (nail polish remover)
COVID-19
Many case reports have documented methemoglobinemia in patients with COVID-19. Most of these patients had taken hydroxychloroquine and were critically ill. [60] It is unclear whether the severe infection leads to methemoglobinemia or the oxidative stress contributes to the severity of COVID-19 infection. A few cases of methemoglobinemia have been reported in patients with COVID-19 with underlying G6PD deficiency, although no precipitating drugs were identified. [61, 62, 63]
Epidemiology
United States statistics
Hereditary methemoglobinemia is rare. The most common cause of congenital methemoglobinemia is cytochrome b5 reductase deficiency (type I b5R). This enzymatic deficiency is endemic in certain Native American tribes (Navajo and Athabaskan Alaskans).
Most cases of methemoglobinemia are acquired and result from exposure to certain drugs or toxins. One of the more common causes of acquired methemoglobinemia is exposure to topical benzocaine during medical procedures. An estimated 0.115% of patients undergoing transesophageal echocardiography (TEE) develop methemoglobinemia. [15, 64, 65]
A large retrospective cohort study found a high incidence of methemoglobinemia (up to 19.8%) in 167 pediatric patients receiving dapsone for PCP prophylaxis. [66] The median methemoglobin level was 9% (range, 3.5-22.4%). The risk of developing methemoglobinemia was increased in those patients receiving a higher dose of dapsone (≥20% above the target dosage of 2 mg/kg/day).
International statistics
Methemoglobinemia occurs rarely throughout the world. Cytochrome b5 reductase deficiency (type I b5R) is also endemic in the Yakutsk people of Siberia.
Age-related demographics
Children, especially those younger than 4 months, are particularly susceptible to methemoglobinemia. The primary erythrocyte protective mechanism against oxidative stress is the NADH system. In infants, this system has not fully matured, and the NADH methemoglobin reductase activity and concentrations are low. Children between the ages of 6 and 10 years may have higher baseline levels of methemoglobinemia than adults do. [67]
Race-related demographics
The congenital form of methemoglobinemia due to cytochrome b5 reductase deficiency (type Ib5R) is endemic in certain ethnic groups. These groups include the Navajo, Athabaskan Alaskans, and the Yakutsk people in Siberia. Because G6PD deficiency is a risk factor for acquired methemoglobinemia, populations in which such deficiency is endemic, including populations of Mediterranean and African descent, are at higher risk for acquired methemoglobinemia.
Prognosis
The prognosis of mild cases of methemoglobinemia is very favorable. In severe cases, the prognosis is determined by the degree of anoxic end-organ damage. Complications of methemoglobinemia may include myocardial infarction, seizure, coma, and death. As methemoglobin levels increase, patients demonstrate evidence of cellular hypoxia. Death occurs when methemoglobin fractions approach 70%. Complications, including death, can occur at lower levels in patients with significant comorbidities.
The clinical course of hereditary forms of methemoglobinemia is generally benign. Patients are usually asymptomatic, except for the presence of chronic cyanosis. However, individuals with type II b5 cytochrome reductase deficiency have a markedly shortened life expectancy, primarily because of multiple neurologic complications.
Acquired methemoglobinemia is usually mild but may be severe and rarely fatal, depending on the cause. Mild-to-moderate transient methemoglobinemia may be present but may escape clinical detection; a high index of suspicion must be maintained. [68]
Patients with acquired methemoglobinemia due to toxin exposure can be severely ill when diagnosed. In some cases, acquired toxic methemoglobinemia can be life-threatening, particularly when the exposure is intentional or the condition is not recognized. However, acquired toxic methemoglobinemia usually responds to treatment when it is recognized and properly treated.
Patient Education
Patients with inherited methemoglobinemia should be counseled regarding the avoidance of toxins, chemicals, and certain drugs (eg, dapsone). Genetic counseling is important. Treatment of type II cases does not prevent or reverse CNS progression.
Patients with both congenital and acquired methemoglobinemia should receive instructions regarding avoidance of precipitating factors.
Patients who develop methemoglobinemia from the oxidant stress of pharmaceutical agents should be warned about other potent oxidant compounds. Patients who develop methemoglobinemia secondary to environmental exposure require a meticulous workup to prevent reexposure to the offending agent. All workplace or household members should be evaluated.
Patients receiving therapy for chronic methemoglobinemia should receive adequate information regarding the risks and benefits expected with treatment.
For patient education information, see Anemia.
-
Note chocolate brown color of methemoglobinemia. In tubes 1 and 2, methemoglobin fraction is 70%; in tube 3, 20%; and in tube 4, normal.



