Background
In the 52 years since the original description of Menkes kinky hair disease (MKHD), advances in understanding the clinical, biochemical, and molecular aspects of this rare disorder of copper metabolism have outstripped progress in the design of effective therapies. [1] The most promising therapy to date, very early subcutaneous copper injections, has normalized neurodevelopmental outcome in some individuals with Menkes kinky hair disease (approximately 30% in the author's experience) and mitigated the neurologic effects in others. However, some patients with Menkes kinky hair disease (nearly 50% in the author's experience) have not derived substantive benefit from this approach, despite very early institution of treatment.
Identification of the Menkes gene by positional cloning has enabled molecular diagnosis of females who carry the gene and at-risk fetuses in certain families, enhancing preventive efforts. Evidence that the gene encodes a highly conserved copper-transporting adenosine triphosphatase (ATPase) has stimulated investigation of the molecule's normal function in prokaryotic and eukaryotic systems. [2] Knowledge gleaned from such efforts may ultimately suggest the novel therapeutic strategies needed to achieve normal neurologic outcomes in patients with Menkes kinky hair disease regardless of mutation severity. Although early recognition of infants with Menkes kinky hair disease prior to neurologic damage remains a fundamental requirement, the recent advances provide a glimmer of hope in efforts to improve matters for individuals with Menkes kinky hair disease and the families who care for them. [3, 4]
History of the disorder
History of this disorder can be traced to as early as 1937, when Australian veterinary scientists recognized the critical role of copper in mammalian neurodevelopment through the association of copper deficiency with demyelinating disease in ataxic lambs. These animals' mothers had grazed in copper-deficient pastures throughout their pregnancies, and their offspring consequently demonstrated symmetric cerebral demyelination and gross pathologic changes, such as porencephalic cyst formation and cavitation.
Based on this connection between copper deficiency and demyelinating disease, neurologists at Oxford in 1948 investigated copper metabolism in a group of patients with multiple sclerosis (MS), a demyelinating disease of adults. Those studies excluded defective copper metabolism as the cause of MS, and Professor David Danks later identified Menkes kinky hair disease as a human example of abnormal myelination due to copper deficiency. [5, 6]
Danks' discovery in 1972 was based on his recognition that the unusual hair of infants with Menkes kinky hair disease appeared similar in texture to the brittle wool of sheep raised on copper-deficient soil in Australia, where wool production remained a major industry. [5, 6] He measured serum copper in 7 patients with Menkes kinky hair disease and found low levels in all 7 individuals. Serum levels of ceruloplasmin, an important copper enzyme, were also subnormal. Thus, observations made 35 years apart concerning the effects of copper deficiency in Australian sheep became extremely relevant to a human inborn error of metabolism.
This important biochemical finding sparked renewed interest in the phenotype that had been delineated meticulously 10 years earlier by John Menkes, MD, and colleagues at Columbia University in New York. [7] Menkes had reported 5 male infants in a family of English-Irish heritage who were affected with a distinctive syndrome of neurologic degeneration, peculiar hair, and failure to thrive. The boys appeared healthy at birth and throughout the first several months of life, but then they experienced seizures and developmental regression and ultimately died when aged 7 months to 3.5 years. The pedigree of the family strongly suggested that the condition was an X-linked genetic disease. Subsequent case reports confirmed that Menkes "kinky hair" disease was a newly recognized syndrome with unique clinicopathologic features.
The association of this disorder with abnormal copper metabolism had a number of important effects. Clinical diagnosis was facilitated by the availability of a reliable biochemical marker (ie, low serum copper and ceruloplasmin). Treatment for a previously fatal disease could be considered by way of copper replacement, and physiologically suitable forms had been reported. Delineation of the basic defect appeared possible, particularly when an excellent mouse model for the human phenotype was recognized and when cultured cells of patients with Menkes kinky hair disease were demonstrated to have distinctive abnormalities in copper handling. The latter findings were applied rapidly as a method of prenatal detection by analysis of cultured amniocytes.
During the following 15 years, additional descriptions of clinical, biochemical, and pathologic features of patients with Menkes kinky hair disease brought attention to the phenotypic spectrum of the disorder. Reports of treatment with copper supplementation in the classic severe type generally indicated little impact on the dismal natural history. A mild form of the disease was noted in which neurologic abnormalities were far less profound. Recognition of a close biochemical relationship between Menkes kinky hair disease and type IX Ehlers-Danlos syndrome (ie, occipital horn syndrome [OHS]) suggested a gene locus comparable to that in the mouse, wherein similar differences in neurologic effects, connective tissue manifestations, and longevity had been reported between 2 apparently allelic variants.
Mapping studies localized the gene to the long arm of the X chromosome close to the centromere. Metallothionein, a copper protein overexpressed in Menkes cultured cells and suspected by some as the primary abnormality, was excluded from direct consideration by localization to chromosome 16 in somatic cell hybrid studies. Experience with prenatal detection increased, and biochemical tests that used chorionic villus samples were developed to enable earlier diagnosis in at-risk pregnancies. A Menkes parents and professionals association was formed in the United States.
In 1987, a female with classic Menkes kinky hair disease caused by an X-autosome chromosomal translocation was reported. This critical observation narrowed the cytogenetic region containing the Menkes locus to Xq13, and cell lines established from this patient ultimately led to cloning of the gene. From a medical perspective, improved outcomes in several patients treated from a very early age with a copper-histidine complex were reported, and a protocol at the National Institutes of Health (NIH) was established to further evaluate the clinical and biochemical effects of this agent in patients with Menkes kinky hair disease.
Identification of the Menkes gene by positional cloning was reported in 1993. [8] This landmark discovery disclosed that the Menkes gene product is a member of a highly conserved family of cation-transporting ATPases [9] , which are molecules that function in the transport of ions across cellular and intracellular membranes. In conjunction with previous data characterizing the biochemical abnormalities in patients with Menkes kinky hair disease and their cultured cells, this finding suggests that the basic defect in Menkes kinky hair disease is the failure of a plasma membrane pump that normally extrudes copper from cells or failure of a pump that normally transports copper into an intracellular organelle such as endoplasmic reticulum.
Thus, in the nearly 40 years since its initial description, Menkes kinky hair disease has been the subject of extensive clinical and scientific scrutiny. The attention culminated in the detection of the faulty gene product, a discovery that provided basic insight into mammalian copper metabolism and presaged a new era in the investigation and history of this disorder.
Animal models of kinky hair disease
The mottled mouse provides an excellent animal model for Menkes kinky hair disease. The mottled and Menkes loci are located in homologous regions of their respective X chromosomes, and several allelic variants have been recognized in the mouse, predicting the possibility of a similar situation in humans. [10]
One of the best studied mottled mutants, the brindled (Mobr) male hemizygote, exhibits decreased coat pigmentation, tremor, general inactivity, death when aged 14 days, increased intestinal copper levels with low levels in the liver and brain, and decreased copper enzyme activities. Of great interest is the observation that healthy viability can be restored in these mutants if a single copper injection is provided during the first week of life, whereas treatment is ineffective when administered later (eg, when aged 12 d).
This response is also characteristic of the macular mouse, a biochemically similar model of Menkes kinky hair disease discovered in Japan. These findings suggest the following: (1) the existence of a critical period in murine neurodevelopment during which copper is essential, and (2) the brindled mutation does not completely impede proper use of copper when the block in intestinal absorption is bypassed.
Male mice hemizygous for other mottled alleles (eg, tortoise, dappled, viable-brindled) also exhibit reduced viability. In contrast, the blotchy mutant (Mo-blo) has healthy viability but more pronounced connective tissue abnormalities. Cultured fibroblasts from all the mutants tested demonstrate the abnormal copper accumulation characteristic of Menkes kinky hair disease.
Biochemical investigation of brindled and blotchy mutants has been extensive. These findings suggest that cytochrome c oxidase (CCO) may be affected more than other copper enzymes in the brindled mutant and that partial restoration of CCO activity in the brain may be responsible for the clinical improvement associated with early copper therapy.
In untreated brindled mice, CCO deficiency has been correlated with progressive neuropathologic changes. In the blotchy mutant, CCO deficiency is less severe than in the brindled mutant, whereas lysyl oxidase (LO) deficiency appears more pronounced, suggesting that the blotchy mutant may be analogous to the human occipital horn phenotype in which connective tissue manifestations predominate. Interestingly, LO response to copper treatment seems better in the brindled mutant than in the blotchy mutant. Also noteworthy is the apparent preservation of normal CCO and superoxide dismutase (SOD) activity in certain organs of both mutants, including the kidney, which is one organ that manifests the copper accumulation phenotype.
Direct measurement of dopamine beta-hydroxylase (DBH) activity in the mottled mutants is complicated by the fact that most assays for DBH require the addition of exogenous copper to samples being measured. The provision of copper presumably circumvents the basis for deficient DBH activity in vivo in tissues of these mutants. Brindled and blotchy mice in which low levels of norepinephrine (NE) in the brain indicated significant DBH deficiency actually demonstrated increased brain DBH when assayed in vitro. These findings suggested that DBH apoenzyme was available in adequate amounts, indeed amounts are perhaps increased in a compensatory manner, but that enzyme function was impaired because of unavailability of copper as a cofactor in vivo. Data on DBH response to copper therapy in the mottled mutants are limited.
Copper/zinc (Cu/Zn) SOD activity is not reduced in either mouse mutant to the same extent as the other copper enzymes studied, nor is its activity enhanced as much (if at all) by copper treatment. In one study of cultured blotchy fibroblasts, measurable SOD activity did not differ from controls. The consistent favorable clinical response to copper treatment in the brindled mutant represents a distinct difference from the experience in most patients with Menkes kinky hair disease.
Cloning of the mottled gene by 2 laboratories (Gitscher, Mercer) and identification of the mutants (ie, brindled, blotchy, dappled) and other alleles by several laboratories (ie, Gitscher, Mercer, Boyd) have improved the understanding of the relationship between mottled phenotype and genotype. Some of these mutant alleles may hold promise for evaluating potential new therapies for Menkes kinky hair disease.
Pathophysiology
As an X-linked disease, Menkes kinky hair disease typically occurs in males who present when aged 2-3 months with loss of previously obtained developmental milestones and the onset of hypotonia, seizures, and failure to thrive. Characteristic physical changes of the hair and facies, in conjunction with typical neurologic findings, often suggest the diagnosis. In 1988, Baerlocher and Nadal compiled the presenting signs and symptoms of 127 patients with Menkes kinky hair disease whose cases had been reported in the medical literature before 1985. [11] The less distinctive appearance of very young infants with Menkes kinky hair disease before the onset of neurodegeneration is discussed separately below. In the natural history of classic Menkes kinky hair disease, death usually occurs by the time the individual with Menkes kinky hair disease is aged 3 years.
Physical presentation
The scalp hair of infants with classic Menkes kinky hair disease is short, sparse, coarse, and twisted. The hair is often less abundant and even shorter on the sides and the back of the head than on the top. The twisted strands may be reminiscent of those in steel wool cleaning pads. The eyebrows usually share the unusual appearance. Light microscopy of patient hair illustrates pathognomonic pili torti (ie, 180° twisting of the hair shaft) and often other abnormalities, including trichoclasis (ie, transverse fracture of hair shaft) and trichoptilosis (ie, longitudinal splitting of shaft). Hair tends to be lightly pigmented and may demonstrate unusual colors, such as white, silver, or grey; however, in some individuals with Menkes kinky hair disease, the hair is pigmented normally.
The face of the individual with Menkes kinky hair disease has pronounced jowls, with sagging cheeks and ears that often appear large. The palate tends to be high-arched, and tooth eruption is delayed. Noisy sonorous breathing is often evident. Although findings on auscultation of the heart and lungs are usually unremarkable, pectus excavatum (chest deformity) is a common thoracic finding. Umbilical and/or inguinal herniae may be present. The skin often appears loose and redundant, particularly at the nape of the neck and on the trunk.
See the image below.
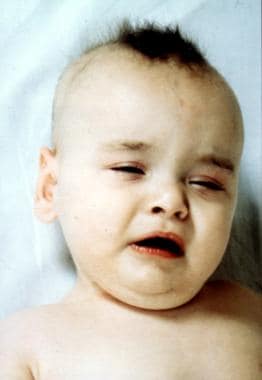 Classic Menkes kinky hair disease in an 8-month-old male infant. Note the abnormal hair, eyelid ptosis, and jowly facial appearance.
Classic Menkes kinky hair disease in an 8-month-old male infant. Note the abnormal hair, eyelid ptosis, and jowly facial appearance.
Neurologically, profound truncal hypotonia with poor head control is invariably present. Appendicular tone may be increased with thumbs held in an adducted cortical posture. Deep tendon reflexes are often hyperactive. The suck and cry are usually strong. Visual fixation and tracking are commonly impaired, whereas hearing is normal. Developmental skills are confined to occasional smiling and babbling in most patients with Menkes kinky hair disease. Growth failure commences shortly after the onset of neurodegeneration and is asymmetric, with linear growth relatively preserved in comparison to weight and head circumference. Clinical diagnostic tests often produce characteristic results (see Workup).
Biochemical phenotype
The biochemical phenotype in Menkes kinky hair disease involves (1) low levels of copper in plasma, liver, and brain because of impaired intestinal absorption, (2) reduced activities of numerous copper-dependent enzymes, and (3) paradoxical accumulation of copper in certain tissues (ie, duodenum, kidney, spleen, pancreas, skeletal muscle, placenta). The copper-retention phenotype is also evident in cultured fibroblasts and lymphoblasts, in which reduced egress of radiolabeled copper is demonstrable in pulse-chase experiments. This constellation of biochemical findings denotes a primary defect affecting copper transport that begins with impaired absorption at the intestinal level and continues with failed utilization and handling of whatever copper is conveyed to other cells in the body.
Certain clinical features of Menkes kinky hair disease can clearly be related to deficient activity of specific copper-requiring enzymes, and one can speculate on the effects that reduced activity of other copper enzymes would produce. Partial deficiency of DBH, a critical enzyme in the catecholamine biosynthetic pathway, is responsible for a distinctively abnormal plasma and cerebrospinal fluid (CSF) neurochemical pattern in patients with Menkes kinky hair disease. In the author's experience, the ratio of a proximal compound in the pathway, (dihydroxyphenylalanine [DOPA]), to a distal metabolite (dihydroxyphenylglycol [DHPG]) provides a better index of DBH deficiency in patients with Menkes kinky hair disease than NE levels alone.
Plasma and especially CSF levels of NE, the direct product of DBH, are maintained relatively well in some patients with Menkes kinky hair disease, presumably because of suitable compensatory mechanisms. Clinical features of patients with Menkes kinky hair disease potentially attributable to DBH deficiency include temperature instability, hypoglycemia, and eyelid ptosis, which are autonomic abnormalities that may result from selective loss of sympathetic adrenergic function. Similar clinical problems have been reported in patients with Riley-Day dysautonomia, in which DBH deficiency has been documented, and/or in patients with congenital absence of DBH.
A copper-dependent enzyme, peptidylglycine alpha-amidating monooxygenase (PAM), is required for removal of the carboxy-terminal glycine residue characteristic of numerous neuroendocrine peptide precursors (eg, gastrin, cholecystokinin, vasoactive intestinal peptide, corticotropin-releasing hormone, thyrotropin-releasing hormone, calcitonin, vasopressin). Failure to amidate these precursors can result in 100-fold to 1000-fold diminution of bioactivity compared with the mature amidated forms. Although deficiency of tyrosinase, a copper enzyme needed for melanin biosynthesis, is considered responsible for reduced hair and skin pigmentation in patients with Menkes kinky hair disease, PAM deficiency may also contribute to this feature through reduced bioactivity of melanocyte-stimulating hormone, an alpha-amidated compound. PAM deficiency may have more important and wide-ranging physiologic effects that contribute to the Menkes phenotype.
Deficient CCO activity is probably a major factor in the neuropathology of Menkes kinky hair disease. Effects on the brain are quite similar to those in individuals with Leigh disease (ie, subacute necrotizing encephalomyelopathy), in whom CCO deficiency is caused by complex IV respiratory chain defects. As in Leigh disease, patients with Menkes kinky hair disease do not have the severe lactic acidemia associated with other complex IV defects. CCO deficiency peripherally probably also contributes to the hypotonia and muscle weakness evident in patients with Menkes kinky hair disease.
Reduced activity of LO, another copper enzyme, also has major clinical consequences in Menkes kinky hair disease. This enzyme normally acts to deaminate lysine and hydroxylysine as the first step in collagen cross-link formation. Decreased LO activity significantly reduces the strength of connective tissue investing numerous organs and tissues. In patients with Menkes kinky hair disease, vascular tortuosity, bladder diverticula, and gastric polyps are all believed to result from LO deficiency.
Deficiency of Cu/Zn SOD in Menkes kinky hair disease may lower protection against oxygen free radicals and theoretically have cytotoxic effects. Localized brain damage due to such oxidant stress has been postulated as the pathogenetic basis of Parkinson disease. Mutations in the Cu/Zn SOD gene on chromosome 21 have been associated with amyotrophic lateral sclerosis, a motor neuron disease of adult onset. The relative contribution of partial SOD deficiency to the neurodegenerative changes in patients with Menkes kinky hair disease is difficult to assign.
Further pathology
Interesting and varied ocular pathology has been reported, including retinal hypopigmentation and vessel tortuosity, macular dystrophy, congenital cataracts, partial optic nerve atrophy and decreased retinal ganglion cells, and microcysts in the pigment epithelium of the iris.
On occasion, thymic atrophy and impaired T-cell function has been demonstrated in patients with Menkes kinky hair disease and warrants investigation in a larger group, given the apparent predisposition to infectious illness in some patients with the syndrome. Decreased T-cell function has been reported in the macular mouse, an animal model of Menkes disease.
Epidemiology
Frequency
United States
Menkes kinky hair disease is a relatively rare condition with incidence estimates ranging from 1 case per 100,000 live births to 1 case in 250,000. Based on the recent number of annual births in the United States (approximately 3.9 million), an estimated 16-40 infants with Menkes kinky hair disease are expected to be born in this country each year. One third of these infants are predicted to be nonfamilial, representing new mutations.
International
Mutations in the Menkes gene occur in all racial and ethnic groups, presumably at the same frequency as occurs in the United States. Therefore, based on recent estimates of annual world births (approximately 135 million per year), an estimated 540-1350 infants with Menkes kinky hair disease are expected to be born each year worldwide.
Mortality/Morbidity
The life span of children with Menkes kinky hair disease cannot be reliably predicted, although most of these children die by the time they are aged 3 years. Pneumonia, leading to respiratory failure, is a common cause of death, although some patients with Menkes kinky hair disease die suddenly in the absence of any apparent acute medical process. The major morbidity associated with Menkes kinky hair disease involves the neurologic, GI, and connective tissue (including vasculature) systems (see Pathophysiology).
Race
No particular racial or ethnic predilection for Menkes kinky hair disease is noted. For X-linked recessive lethal traits, such as in individuals with Menkes kinky hair disease, genetic theory suggests that one third of infants with Menkes kinky hair disease represent new mutations. Such de novo mutations are expected to occur at equal frequency among all Homo sapiens racial and ethnic groups.
Sex
Menkes kinky hair disease affects males nearly exclusively because it is an X-linked recessive trait. Female carriers generally do not manifest symptoms unless unusual genetic circumstances are present. These include unfavorable lyonization due to skewed X-inactivation, balanced chromosomal translocations with breakpoints lying within the Menkes gene, or sex chromosome aneuploidy (ie, Turner syndrome ([45, XO karyotype]) with a Menkes gene mutation on the sole X chromosome).
Age
As noted above, individuals with Menkes kinky hair disease typically present when aged 6-8 weeks, with parents noticing a delay in developmental progress or the appearance of unusual eye or extremity movements suggestive of seizure activity.
-
Classic Menkes kinky hair disease in an 8-month-old male infant. Note the abnormal hair, eyelid ptosis, and jowly facial appearance.
-
Adolescent patient with typical occipital horn syndrome. Note elbow dislocations and genu valgum. Radiographs exhibited bilateral occipital exostoses of the skull and club-shaped distal clavicles.
-
Successfully treated classic Menkes kinky hair disease. Diagnosis at birth enabled copper therapy to begin when the infant was aged 8 days. The child walked independently when aged 14 months. This patient's mutation (IVS8,AS,dup5) was associated with a transcript harboring a small in-frame deletion, potentially encoding a functional copper adenosine triphosphatase (ATPase).
-
Menkes kinky hair disease copper adenosine triphosphatase (see text for detailed discussion).








