Practice Essentials
Medulloblastoma is the most common malignant brain tumor in children, accounting for up to 25% of primary CNS neoplasms and approximately 50% of all posterior fossa tumors. [1] It is a high-grade (WHO grade IV) embryonal neuroepithelial tumor that arises in the cerebellum and has a tendency to disseminate via the cerebrospinal fluid (CSF). See the image below.
Signs and symptoms of pediatric medulloblastoma
Most patients initially present with the following symptoms:
-
Headaches
-
Emesis
-
Lethargy
Infants may present with irritability, anorexia, and developmental delay.
Later symptoms may include the following:
-
Worsening ataxia involving the lower extremities
-
Diplopia and other cranial nerve findings, such as facial weakness, tinnitus, hearing loss, head tilt, and stiff neck
Funduscopic evaluation may reveal papilledema or optic pallor in infants. Palsy of cranial nerve VI that results in the inability to abduct one or both eyes is common.
See Presentation for more detail.
Diagnosis of pediatric medulloblastoma
Laboratory studies
The routine pretreatment laboratory evaluation for medulloblastoma includes a complete blood cell (CBC) count, measurement of electrolyte levels, and liver and renal function tests. Baseline thyroid function studies and viral titers are also recommended.
Imaging studies
Diagnosis is usually made by magnetic resonance imaging (MRI) or computed tomography (CT).
See Workup for more detail.
Management of pediatric medulloblastoma
With aggressive surgery, craniospinal radiotherapy, and chemotherapy, more than 50% of children with medulloblastoma can be expected to be free of disease 5 years later. Using current treatments, 80-90% of those without disseminated disease can be cured; however, treatment for this disease often results in significant long-term neurologic, endocrinologic, and intellectual sequelae.
See Treatment and Medication for more detail.
Pathophysiology
Medulloblastoma is a heterogeneous disease. Recent advances in understanding the molecular characteristics of medulloblastoma cells allowed for sub-typing based on abnormalities seen at the molecular level. These subgroups are defined by their unique clinical behavior and outcomes.
The WNT-subgroup, which accounts for 10% of medulloblastoma cases in children, is characterized by aberrant activation of the Wingless (WNT) signaling pathway. The most frequently mutated genes in WNT medulloblastoma are CTNNB1 and TP53.
The SHH-subgroup accounts for 30% of medulloblastoma cases in children and is characterized by aberrant activation of the Sonic hedgehog (SHH) signaling pathway. The most frequently mutated genes in SHH medulloblastoma are PTCH1, SMO, SUFU, GLI1 and TP53.
Groups 3 and 4 which accounts for 25 and 35% of medulloblastoma cases respectively, lack involvement of any clearly defined signaling pathway. Common genetic alterations involves MYC, OTX2 and SMARCA4 in group 3, and KDM6A and MYCN in group4.
The most recent WHO classification of medulloblastoma is as follows [2] :
-
Medulloblastoma, genetically defined.
- Medulloblastoma, WNT-activated.
- Medulloblastoma, SHH-activated and TP53-mutant.
- Medulloblastoma, SHH-activated and TP53-wildtype.
- Medulloblastoma, non-WNT/non-SHH.
- Medulloblastoma, group 3.
- Medulloblastoma, group 4.
-
Medulloblastoma, histologically defined.
- Medulloblastoma, classic.
- Medulloblastoma, desmoplastic/nodular.
- Medulloblastoma with extensive nodularity.
- Medulloblastoma, large cell/anaplastic.
-
Medulloblastoma, NOS.
Etiology
Environmental causes
Epidemiological studies investigating parental occupational exposures, environmental exposures, and maternal nutritional intake have not proven a direct link between such factors and the development of childhood brain tumors.
Familial and heritable cancer predisposition syndromes associated with medulloblastoma
In one study, 5 of 37 (13.5%) patients with medulloblastoma were found to have germline mutations in one of the known cancer predisposing genes. [3]
Syndromes known to be associated with medulloblastoma include the following:
-
Turcot syndrome (related to germline mutations in APC)
-
Rubinstein-Taybi syndrome (related to germline mutations in CREBBP)
-
Gorlin syndrome (associated with germline PTCH1 and SUFU mutations)
-
Li-Fraumeni syndrome (associated with germline mutations in TP53)
-
Fanconi anemia
Epidemiology
United States statistics
Approximately 250 new patients are diagnosed annually.
International statistics
Exact figures are unknown. In general, brain tumors occur at a rate of 2.5-4 per 100,000 at-risk children per year. Of these, approximately 18% are medulloblastoma.
Race-, sex-, and age-related demographics
No racial predisposition is noted. Data from the Surveillance, Epidemiology, and End Results (SEER) program showed that patients aged 0-14 years in the United States have an incidence rate per million population of 5.7 in whites and 5 in blacks. [4]
US incidence per 1 million population for patients aged 0-14 years is 6.1 for boys and 4.5 for girls.
Peak age of incidence is during the first decade of life. Approximately 80% of patients are diagnosed in the first 15 years of life.
Prognosis
Prognostic factors include the following:
- Age at diagnosis
- Metastatic stage (M stage) at time of presentation
- Extent of surgical resection
- Histology
- Molecular subtype
The specific risk groups based on clinical findings and morphology are defined as follows:
-
Average-risk disease: This risk group is defined as patients older than 3 years who have no evidence of dissemination and with less than 1.5 cm 2 of residual tumor postoperatively. The 5-year survival rate for this group is currently 85%.
-
High-risk disease: This risk group is defined as patients older than 3 years with evidence of metastatic disease, with more than 1.5 cm 2 of residual tumor postoperatively and/or large cell/anaplastic histology. Large cell/anaplastic histology is commonly associated with unfavorable molecular changes like MYC amplification and predicts poor outcome. The 5-year survival rate for this group is currently 30-60%.
-
Infants: This group is defined as patients younger than 3 years. The 5-year survival rate is 30-70% depending on clinical and molecular risk factors; patients with metastatic disease do considerably worse while those with WNT-activated or SHH-activated medulloblastoma are much more likely to survive.
The following genetically defined subgroups have variable outcomes:
-
Patients with WNT-activated medulloblastoma have excellent prognosis with more than 90% long-term survival.
-
Patients younger than 4 years with SHH-activated medulloblastoma do well with 5-year event free survival of 85% when treated with surgery and chemotherapy alone. Children older than 4 years with SHH-activated medulloblastoma without TP53 mutation do fairly well with 5-year event free survival of 60%. However, patients with SHH-subgroup with TP53 mutation do significantly worse.
-
Patients with Group 3 have the worst outcome with close to 50% long-term survival.
-
Patients with Group 4 have close to 75% long-term survival.
Morbidity/mortality
Clinical risk group stratification is continuing to evolve but is currently based on four principal features including age, extent of postoperative residual disease, and the metastasis stage.
Molecular subtype provides significant information regarding tumor behavior and response to treatment. Patients with WNT subtype for example have an excellent prognosis, while patients with MYC or MYCN amplification do poorly.
Despite successful treatment, a significant number of patients have neurocognitive, neurologic and endocrinologic deficits. Many children subsequently develop learning difficulties that require individualized educational programs. Biochemical growth deficiency is observed in 70-80% of patients, and some degree of growth impairment is present in well over half of patients after treatment. Thyroid and gonadotropin hormonal deficiency may also occur. Craniospinal radiation, a mainstay of treatment, has been implicated as a major cause of these deficits.
Complications
The following complications can be classified based on their etiology:
Disease-related
-
Temporary or permanent neurologic impairment, particularly cerebellar dysfunction
-
Headaches
Surgical complications
-
Posterior fossa syndrome (mutism, cerebellar dysfunction, cranial nerve palsy, and hemiparesis; generally temporary)
-
Bleeding
-
Shunt malfunction or infection
Radiation-induced
-
Nausea/vomiting, anorexia
-
Other GI toxicity (e.g. mouth sores, diarrhea)
-
Cytopenias (from irradiation of vertebral bodies, which encompasses 40% of bone marrow) including lymphopenia
-
Neurocognitive impairment
-
Somnolence syndrome (transient sleepiness, typically occurs 4 to 6 weeks from start of radiation)
-
Radiation necrosis, a potential long-term (may develop months or even years after) sequela resulting in edema from death of tumor cells or tissue from radiation exposure
-
Intratumoral hemorrhage
-
Ototoxicity
-
Hypopituitarism (e.g. hypothyroidism, growth hormone deficiency)
-
Secondary malignancies to radiation field areas
Chemotherapy-related
-
Anemia causing headaches and fatigue
-
Thrombocytopenia and risk for bleeding
-
Neutropenia and increased risk for infections
-
Mucositis (can be especially severe with thiotepa)
-
Constipation (vincristine-related)
-
Organ toxicity
Hepatotoxicity
Nephrotoxicity, ototoxicity with cisplatin/carboplatin
Hemorrhagic cystitis with cyclophosphamide
Neurotoxicity with vincristine
-
Secondary leukemia from alkylator therapy
-
Metabolic syndrome (diabetes, hyperlipidemia and heart disease) in stem cell transplant recipients
-
Infertility
Patient Education
Patients and family members should be instructed about the care of the central venous catheter.
Patients should be instructed about measures to minimize infections (eg, proper handwashing, central line care, avoiding sick contacts) and seeking immediate medical attention for fevers or other signs of infection that manifest during therapy.
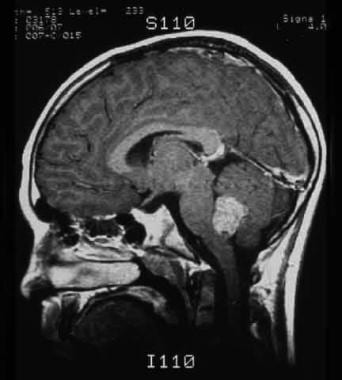
-
MRI showing a medulloblastoma of the cerebellum.
-
Section displaying Homer-Wright rosettes and pseudorosettes of a medulloblastoma.
-
This section displays a typical medulloblastoma, composed of undifferentiated cells with deeply basophilic nuclei of variable size and shape and little discernible cytoplasm.
Tables
Tumor Stage |
|
T1 |
tumor < 3 cm in diameter |
T2 |
tumor ≥3 cm in diameter |
T3a |
tumor >3 cm and with extension into Aqueduct of Sylvius or foramen of Luschka |
T3b |
tumor >3 cm and with unequivocal extension into brainstem |
T4 |
tumor >3 cm with extension past Aqueduct of Sylvius or down past foramen magnum |
Metastases Stage |
|
M0 |
no evidence of gross subarachnoid or hematogenous metastasis |
M1 |
microscopic tumors cells found in CSF |
M2 |
gross nodular seeding intracranially beyond the primary site (in cerebellar/cerebral subarachnoid space or in third or lateral ventricle) |
M3 |
gross nodular seeding in spinal subarachnoid space |
M4 |
metastasis outside cerebrospinal axis |









