Background
The study of fatty acid metabolism gained importance during the 1970s when investigators and clinicians recognized patients who appeared to have genetic defects in this area. In 1973, carnitine palmitoyltransferase (CPT) deficiency became the first fatty acid metabolism condition to be defined. With attention focused on the definition of additional disorders, researchers described patients with a Reye syndrome–like presentation who excreted dicarboxylic acids of chain lengths C6-C10 in their urine.
In 1976, Gregersen and colleagues described a patient with similar findings and theorized that a beta-oxidation defect was responsible; thus, expectations were raised that this type of defect would soon be identified. [1] By 1982, at least 2 reports of patients thought to suffer from defects in beta-oxidation were published. In 1983, Gregersen et al demonstrated a medium-chain acyl-coenzyme A (CoA) dehydrogenase (MCAD) deficiency in a patient with hypoketotic hypoglycemia. [2] Stanley and colleagues described several children with similar clinical presentations who were MCAD deficient and confirmed the demonstration in the same year. [3]
The clinical entity known as MCAD deficiency was biochemically defined less than 35 years ago; however, some believe the condition to be at least as common in newborns as phenylketonuria, with an incidence approximating 1 per every 12,000 live births. A recent report from Europe indicates an incidence in Bavaria of 1:8456 in more than 500,000 newborns screened. [4]
Pathophysiology
The beta-oxidation cycle permits the cell to extract energy from the breakdown of fatty acids with linkage to an accessory pathway for the formation of acetoacetate. Beta-oxidation is a complex mitochondrial pathway that is dependent on the presence of adequate cytosolic carnitine and 2 mitochondrial membrane-bound enzymes: CPT I and CPT II.
In the cytosol, a saturated, straight-chain fatty acid molecule with no double bonds is activated by the action of fatty acyl-CoA synthetase to form its corresponding acyl-CoA. This acyl-CoA is linked to carnitine by the action of CPT I, with simultaneous transport across the mitochondrial membrane barrier. Once inside the mitochondrion, the action of CPT II at the inner surface of the membrane releases free carnitine, which exits to the cytosol and leaves behind the acyl-CoA molecule.
Beta-oxidation cycle
Entry into the beta-oxidation cycle requires the action of acyl-CoA dehydrogenase, the first enzyme in the sequence, which removes electrons from the alpha-carbon and the beta-carbon, introducing a double bond. The electrons are transferred to the flavin cofactor essential for normal enzyme activity. These are, in turn, transferred to the electron transport chain with the production of ATP.
The next step is the introduction of a water molecule and resaturation of the double bond to form fatty enoyl-CoA.
Oxidation of the hydroxyl substituent group on the beta-carbon creates an inherently unstable beta-ketoacyl-CoA compound. In the process, another electron transfer occurs, this time to nicotinamide-adenine dinucleotide (NAD), and more ATP is produced by passage down the electron transport chain.
Cleavage of the 3-keto compound at the now unstable alpha-beta carbon bond and transfer of another CoA moiety to the new fragment results in 2 products: acetyl-CoA, composed of the carbonyl and original alpha-carbon from the starting molecule, and a new fatty acyl-CoA that is 2 carbons shorter than the original molecule.
In addition to its intrinsic importance in the use of alternative fuels, the process of beta-oxidation clearly illustrates the role of vitamin cofactors in metabolism. Both riboflavin and nicotinamide are key to the ferrying of electrons to the cytochromes for production of ATP, without which the breakdown of fatty acid would be utterly useless to the cell's energy economy.
The following 2 additional points are noteworthy regarding beta-oxidation:
-
Fatty acids shorter than C12 do not require CPT activity for mitochondrial entry.
-
At least 3 separate acyl-CoA dehydrogenases are known; they are as follows:
Long-chain acyl-CoA dehydrogenase (Length of fatty acid greater than C12)
Medium-chain acyl CoA dehydrogenase (Length of fatty acid C6-C12)
Short-chain acyl-CoA dehydrogenase (Length of fatty acid less than C6)
Fatty acids longer than C12 can be oxidized by beta-oxidation down to a 12-carbon fatty acyl-CoA; those shorter than C6 are also normally oxidized.
The pathophysiology of MCAD deficiency results from the inability to carry out the first step of beta-oxidation. The molecular implication of most mutations in this disorder is a loss of enzymatic function due to protein misfolding; the amino acid substitutions secondary to the genetic mutations impairs the acquisition of a normal 3-dimensional shape. [5, 6, 7] Any clinical situation in which fatty acid oxidation is required, such as fasting or metabolic stress due to illness, results in continued glucose consumption and a markedly reduced or absent corresponding increase in ketone body production. A 2016 study has suggested that metabolic stress causing flooding of the β-oxidative pathway with substrate may contribute to competitive inhibition, thus enhancing metabolic decompensation. [8]
The ultimate clinical result is severe hypoglycemia and hypoketonuria with accumulation of monocarboxylic fatty acids and dicarboxylic organic acids, which are structural analogues of the fatty acids that cannot pass through the MCAD step. These dicarboxylic acids include adipic (C6), suberic (C8), sebacic (C10), and dodecanedioic (C12). Each is formed by an alternative metabolic pathway called Ω-oxidation that attempts, without success, to begin oxidation at the opposite end of the fatty acid. These omega-oxidation products appear in urine; an appropriately equipped laboratory can identify them and a diagnosis can be expeditiously made. As in propionic acidemia, the cell attempts to conserve free CoA by substitution with carnitine, with a resultant urinary excretion of acyl-carnitine compounds.
Octanoic acid (a C8 fatty acid), which accumulates during an impending metabolic decompensation in an affected patient, is a well-known mitochondrial toxin; this may account for the disruption of ammonia metabolism that often accompanies the clinical presentation of MCAD deficiency. In addition, octanoate has also been shown to reduce oxidation of glucose by rat cerebral homogenates by 70%, which may significantly contribute to the neurological abnormalities. The hypoglycemia and hyperammonemia combine to account for the lethargy and coma that culminate in cerebral edema if left untreated.
Finally, gluconeogenesis is effectively disabled in MCAD deficiency because it depends on the activity of pyruvate carboxylase to produce oxaloacetate, a reaction that is downregulated by diminished mitochondrial acetyl-CoA. Consequently, gluconeogenesis cannot compensate for the continuing consumption of existing glucose and the inability to shift to oxidation of alternative fuels, specifically fatty acids.
Epidemiology
Frequency
United States
Inherited as an autosomal recessive trait, MCAD deficiency was found in at least one series, which used a population-screening technique, to occur in approximately 1 in every 8500 live births. [4]
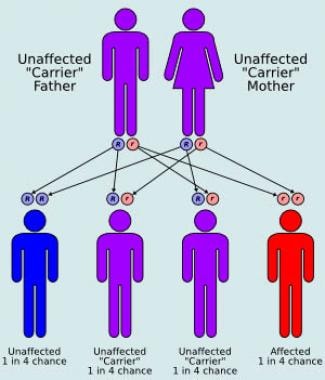 Autosomal recessive inheritance.
Autosomal recessive inheritance.
This figure seems somewhat high, but the true incidence rate is almost certainly among the highest of the inborn errors of metabolism, rivaling that of phenylketonuria. In part because of the supposed frequency of the disorder, nationwide debate has resulted in many states adding MCAD to their existing newborn metabolic screening programs. Although screening is feasible, each of the available methodologies has drawbacks. Tandem mass spectrometry requires a very large capital investment in instrumentation, whereas molecular probes are not yet commercially available for all mutations that result in clinical disease. Many additional states are likely to add MCAD deficiency soon to their routine newborn metabolic screening program, and the true incidence of the disease in the United States will be revealed.
International
The application of tandem mass spectrometry to newborn metabolic screening in many other countries throughout the world has supplied a better understanding of the worldwide incidence of MCAD deficiency. [9, 10] The average incidence rate among more than 8 million babies was 1 per 14,600 live births, with a range of 1 per 13,500 to 1 per 15,900. [11] A single mutation accounted for more than 50% of all diagnosed cases.
Mortality/Morbidity
Some authors note that MCAD deficiency may be a cause of sudden infant death syndrome (SIDS), a concept that has largely been discredited by extensive postmortem study over the past decade. The tendency for development of very severe hypoglycemia is associated with a risk of serious damage to the CNS and other organs. The possibility of cerebral edema and coma leading to death is a cause of legitimate concern.
Sex
Because the mutation is an autosomal recessive trait, equal gender distribution is anticipated.
Age
Because MCAD deficiency is a genetically transmitted disorder, it is present from conception. Clinical onset can occur at any time during infancy; however, the tendency is for onset in infants aged 3 months and older, when overnight feedings begin to diminish in frequency. One report suggests the potential for neonatal decompensation associated with compound heterozygosity of a c.199T>C mutation and the common c.985A>G mutation. [12] The increase in fasting time exposes the underlying defect, which leads to the clinical presentation.
Early diagnosis is imperative because serious morbidity and mortality is associated with initial onset in more than 25% of undiagnosed individuals. A recent report [13] suggests that residual enzyme activity of greater than 10% of normal is either sufficient for most metabolic needs, and/or may delay onset and determine clinical severity. This observation deserves greater attention in order to expedite clinical prognosis.
As is the case in many other inherited metabolic disorders, adult-onset MCAD has been reported as well. The individuals reported have been previously healthy adults. Although reports of affected adults are not surprising because of the high incidence rate of the disease, the absence of symptoms prior to clinical onset is surprising. A recent report of a newborn screened out as abnormal led to identification of the asymptomatic mother as an affected individual. [14]
-
Autosomal recessive inheritance.






