Practice Essentials
May-Hegglin anomaly (MHA) is an autosomal dominant disorder characterized by various degrees of thrombocytopenia that may be associated with purpura and bleeding; giant platelets containing few granules; and large, well-defined, basophilic, cytoplasmic inclusion bodies in granulocytes that resemble Döhle bodies (see the image below). [1, 2, 3] MHA is one of a family of macrothrombocytopenias characterized by mutations in the MYH9 gene. [4, 5] The other members of this family include Sebastian syndrome, [6] Epstein syndrome, [7] and Fechtner syndrome, [8] although it is likely, as MYH9-related disorders are better characterized, that these time-honored eponyms will fade away.
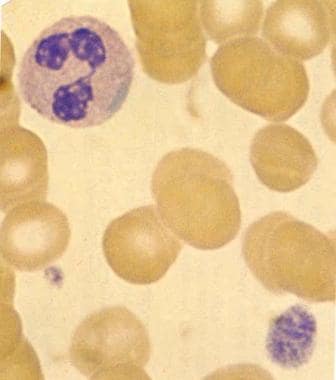 Blood smear (original magnification ×2000) in patient with May-Hegglin anomaly (MHA) demonstrates characteristic giant platelet with poorly defined granulation. Normal-sized platelet is also present. Trilobed neutrophil contains large, well-defined, basophilic, peripherally placed cytoplasmic inclusion body (resembling Döhle body). Image used with permission from Little, Brown.
Blood smear (original magnification ×2000) in patient with May-Hegglin anomaly (MHA) demonstrates characteristic giant platelet with poorly defined granulation. Normal-sized platelet is also present. Trilobed neutrophil contains large, well-defined, basophilic, peripherally placed cytoplasmic inclusion body (resembling Döhle body). Image used with permission from Little, Brown.
Signs and symptoms
Patients are often asymptomatic. The bleeding tendency associated with MHA is generally mild and is thought to mainly depend on the degree of thrombocytopenia. Symptoms of bleeding can include the following:
-
Recurrent epistaxis
-
Gingival bleeding
-
Easy bruising
-
Menorrhagia
-
Excessive bleeding associated with surgical procedures
-
Postpartum hemorrhage
Physical findings are often normal. Findings of abnormal bleeding may be subtle and may include the following:
-
Bruising
-
Petechiae
-
Active bleeding from mucosal surfaces
-
Prolonged and excessive bleeding and oozing associated with lacerations and sutures
It is important to look for associated clinical features of other MYH9-related disorders (ie, Sebastian syndrome, Epstein syndrome, Fechtner syndrome). The following findings may be noted in these syndromes, which may overlap depending on the underlying mutation.
-
High-frequency hearing loss
-
Cataracts
-
Hematuria
-
Proteinuria
See Presentation for more detail.
Diagnosis
Laboratory studies that may be helpful include the following:
-
Complete blood count (CBC)
-
Assessment of platelet size, volume, and morphology
-
Peripheral blood smear
-
Immunocytochemistry of leukocytes (demonstrating NMMHCIIA complexes, for confirmation)
-
Bleeding time
See Workup for more detail.
Management
Most patients with MHA do not appear to have clinically significant bleeding problems, and specific treatment is not required. The following points may be considered:
-
Corticosteroids and splenectomy are ineffective
-
In rare patients with severe bleeding, platelet transfusion may be required
-
Bleeding risk is not significantly increased by normal vaginal delivery
-
For patients scheduled to undergo surgery, intravenous desmopressin acetate (DDAVP) may be valuable; routine prophylactic platelet transfusions are not usually indicated, but platelets should be kept available
-
Depending on circumstances, refraining from participation in contact or collision sports may be prudent
See Treatment and Medication for more detail.
Background
In 1909, May described the presence of leukocyte inclusions and large platelets in an asymptomatic young woman. In 1945, Hegglin described a man and his 2 sons who were healthy but had a triad consisting of thrombocytopenia, giant platelets, and leukocyte inclusions. This diagnostic triad was later given the eponym May-Hegglin anomaly (MHA). [9]
MHA is an autosomal dominant disorder characterized by various degrees of thrombocytopenia that may be associated with purpura and bleeding; giant platelets containing few granules; and large (2-5 µm), well-defined, basophilic, cytoplasmic inclusion bodies in granulocytes that resemble Döhle bodies. [1]
MHA is one of a family of macrothrombocytopenias characterized by mutations in the MYH9 gene. [4] The other members of this family include Sebastian syndrome, [6] Epstein syndrome, [7] and Fechtner syndrome. [8]
Most patients with MHA do not have clinically significant problems with bleeding and therefore do not require treatment.
Pathophysiology
Patients with MHA have a mutation of the MYH9 gene present in chromosomal region 22q12-13. [10, 4] The mutation results in disordered production of nonmuscle myosin heavy-chain type IIA, which leads to invariable macrothrombocytopenia secondary to defective megakaryocyte maturation. [11] Platelet function in patients with MHA has been reported as normal. [12, 13] ; however, in one study, epinephrine response was described as abnormal in 8 of 15 patients. [14]
Leukocyte Döhlelike inclusion bodies are visualized on standard Wright stain and appear bright blue and spindle-shaped. Ultrastructural studies reveal that these bodies consist of clusters of ribosomes oriented along parallel myosin heavy-chain filaments 7-10 nm in diameter. [15] Neutrophil function is considered to be normal, and patients have no increased susceptibility to infections.
Etiology
May-Hegglin anomaly is one of a family of macrothrombocytopenias characterized by mutations in the MYH9 gene, which is present at chromosomal region 22q12-13 and codes for nonmuscle myosin heavy-chain IIA. [4] The Döhlelike leukocyte inclusions in MHA are due to precipitation of myosin heavy chains in leukocytes.
Analysis of more than 70 families confirms that mutations in MYH9 can lead to May-Hegglin anomaly, Sebastian syndrome, Fechtner syndrome, or Epstein syndrome. [16] In one fifth of individuals, the mutation may sporadically arise. [17] Macrothrombocytopenia is invariably present (see the Table below). [18] The clinical description of these syndromes predated the discovery of the MYH9 gene mutations. Although the theory was that genotype-phenotype correlations would be found, this has not been the case overall. [19] The MYH9 R702 mutation is reportedly associated with the smaller neutrophil inclusions seen in Sebastian syndrome, Fechtner syndrome, and Epstein syndrome. [20] In addition MYH9 exon 24 indel mutations may be associated with nephritis, deafness, and congenital cataracts. [21]
Table. Clinical Features of MYH9 -Related Thrombocytopenias (Open Table in a new window)
Condition |
Macrothrombocytopenia |
Granulocyte inclusions |
Nephritis and Deafness |
Cataracts |
MHA |
Yes |
Linear Döhlelike |
No |
No |
Epstein syndrome |
Yes |
Absent or faint |
Yes |
No |
Fechtner syndrome |
Yes |
Spherical granules |
Yes |
Yes |
Sebastian syndrome |
Yes |
Spherical granules |
No |
No |
Epidemiology
MHA is a rare autosomal dominant disorder. In one review, 180 cases had been reported in the literature. [14] Kindreds have been reported from Italy, France, Germany, and North America. [4] MHA was reported in 15 families in Japan in 1993. [22] The exact incidence of the syndrome is unknown.
Prognosis
The rarity of MHA has led to conflicting literature regarding the risk for bleeding. Asymptomatic patients have been described [12, 13] ; however, abnormal bleeding has also been documented. [14] The bleeding risk is increased by taking drugs that decrease platelet function. The risk for excess bleeding with surgical procedures is unclear. [23] Rare reports have described arterial thrombotic events associated with May-Hegglin anomaly, though the risk remains unclear. [24]
Patient Education
Individuals with MHA should be informed regarding their personal risk of bleeding. They should be made aware that their bleeding risks are associated with the degree of thrombocytopenia.
In patients who are scheduled to undergo surgical procedures or have sustained trauma, the diagnosis of MHA must be discussed because special precautions and procedures may be required to prevent bleeding complications.
Individuals with MHA should be educated to avoid drugs (eg, aspirin) that can adversely affect platelet function.
-
Blood smear (original magnification ×2000) in patient with May-Hegglin anomaly (MHA) demonstrates characteristic giant platelet with poorly defined granulation. Normal-sized platelet is also present. Trilobed neutrophil contains large, well-defined, basophilic, peripherally placed cytoplasmic inclusion body (resembling Döhle body). Image used with permission from Little, Brown.
Tables
Condition |
Macrothrombocytopenia |
Granulocyte inclusions |
Nephritis and Deafness |
Cataracts |
MHA |
Yes |
Linear Döhlelike |
No |
No |
Epstein syndrome |
Yes |
Absent or faint |
Yes |
No |
Fechtner syndrome |
Yes |
Spherical granules |
Yes |
Yes |
Sebastian syndrome |
Yes |
Spherical granules |
No |
No |








