Practice Essentials
Mandibulofacial dysostosis, [1] also known as Treacher Collins syndrome (TCS; entry 154500 in the Online Mendelian Inheritance in Man [OMIM] classification system), is an inherited developmental disorder with a prevalence estimated to range between 1 in 25,000 and 1 in 50,000 live births. [1, 2, 3, 4, 5] Growth of craniofacial structures derived from the first and second pharyngeal arch, groove, and pouch is diminished symmetrically and bilaterally. The condition is recognizable at birth and can also be diagnosed prenatally based on ultrasonography findings. [6] The following images are examples of the characteristic features of this condition.
Management of this condition is lengthy and requires a multidisciplinary approach focused on treatment of symptoms.
This syndrome was named after the eminent British ophthalmologist Edward Treacher Collins (1862-1932), who described the essential features of this syndrome in a paper in 1900. However, some features of this syndrome were probably first described by Thomson and Toynbee in 1846-1847 and later by Berry (1889), who is usually given credit for its discovery. [2] On the European continent, a more common name for this condition is Franceschetti-Zwahlen-Klein syndrome, based on extensive studies of mandibulofacial dysostosis published by the Swiss ophthalmologist Franceschetti and the geneticist Klein (1949).
Signs and symptoms of Treacher Collins syndrome
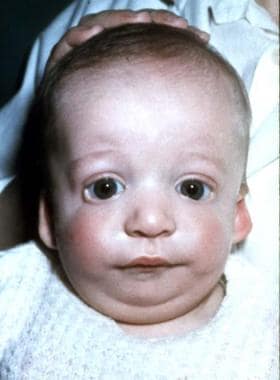
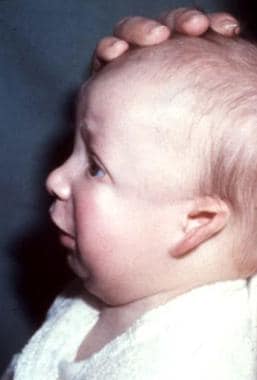
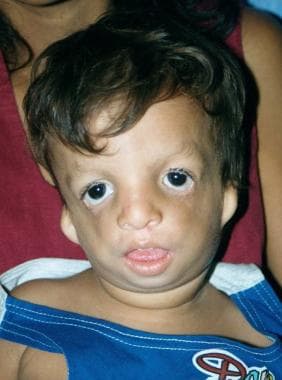
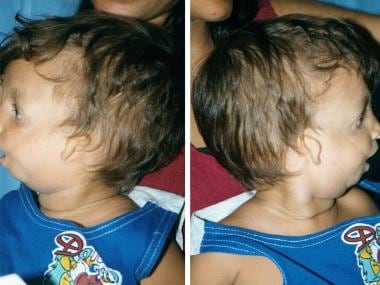
In persons with Treacher Collins syndrome, the nose is of normal size; however, it appears large because of hypoplastic supraorbital rims and hypoplastic zygomas.
The palpebral fissures are downward sloping; the cheekbones are depressed; the pinnae are malformed, with widely varying severity; and the chin recedes with a large, downturned mouth.
In the ears, the pinnae are often malformed, crumpled forward, or misplaced toward the angle of the mandible.
A cleft palate is found in one third of patients with Treacher Collins syndrome, and congenital palatopharyngeal incompetence (foreshortened, immobile, or absent soft palate; submucous cleft palate) is found in an additional one third of patients. [7]
The patient usually has normal intelligence.
Workup in Treacher Collins syndrome
Midtrimester ultrasonography can detect facial dysmorphology. [8, 9]
The following imaging studies should be obtained to visualize craniofacial dysmorphology in detail and repeated, as needed, for surgical planning:
-
Anteroposterior and lateral cephalography
-
Full craniofacial computed tomography (CT) scanning (axial and coronal slices from the top of the skull through the cervical spine)
-
As follow-up, CT scans from orbits through mandible (usually enough for surgical planning)
-
Panoramic radiography
-
Brain magnetic resonance imaging (MRI) for inner auditory canal (IAC) study preferred (If MRI is not available, CT scan may be obtained for IAC.)
Management of Treacher Collins syndrome
In patients with severe manifestations of Treacher Collins syndrome in which airway inadequacy is the prominent feature after birth, a tracheostomy is performed (and may remain for several years, until the lower jaw has sufficiently grown or until alveolar distraction is performed to enable passage of air through the oral cavity). [10]
In patients with severe swallowing difficulty, introduce feeding by gavage or even through a gastrostomy tube, to ensure adequate caloric intake and hydration.
Fit hearing aids shortly after birth if the patient has substantial conductive hearing loss.
Operative repair in Treacher Collins syndrome is based on the anatomic deformity, and the timing of corrections depends on physiologic need and development.
Distraction osteogenesis, an orthopedic method of lengthening bone, has been used to lengthen the neonatal mandible.
For minor obstructions that do not require a tracheostomy and can be corrected with positioning, a tongue-lip adhesion can be considered.
The lateral coloboma of the lower eyelid has traditionally been corrected with a skin-muscle flap from the upper lid or brow.
Macrostomia, if present, can be repaired at the same time with Z-plasty or straight-line skin repair. Cleft palate, present in one third of cases, is typically repaired at approximately age 10-12 months.
Microtia is addressed at age 5-7 years, which is when the external ear is approximately 80-90% of adult size and rib cartilage is of sufficient volume to use as graft material.
Addressing the hard tissues is usually deferred until skeletal maturity. Osteotomies and bone grafts address the long midface, short mandible, and lateral facial clefts.
Epidemiology
Frequency
United States and international
Prevalence of Treacher Collins syndrome is in the range 1 per 25,000 to 1 in 50,000 live births. [2]
Race
Treacher Collins syndrome has no race predilection.
Sex
Males and females are equally affected.
Age
In the vast majority of cases, Treacher Collins syndrome is clearly diagnosed at birth. Because of typical facial dysmorphology in severe cases, it may also be diagnosed prenatally by ultrasonography. In mild cases, with minimal expression of facial features, the syndrome may be undiagnosed at birth.
-
Anteroposterior view of 2-month-old boy with Treacher Collins syndrome.
-
Lateral view of 2-month-old boy with Treacher Collins syndrome.
-
Anteroposterior view of 2-year-old boy with Treacher Collins syndrome.
-
Lateral views of 2-year-old boy with Treacher Collins syndrome.
-
Anteroposterior view of 19-week-old fetus with Treacher Collins syndrome. Diagnosis was confirmed based on fetoscopy images. Tolarova M, Zwinger A. The use of fetoscopy by inborn morphological anomalies. Acta Chir Plast. 1981;23(3):139-51.
-
Lateral view of 19-week-old fetus with Treacher Collins syndrome. Diagnosis was confirmed based on fetoscopy images. Tolarova M, Zwinger A. The use of fetoscopy by inborn morphological anomalies. Acta Chir Plast. 1981;23(3):139-51.
-
Right-lateral cranial radiograph of an 18-month-old infant with Treacher Collins syndrome. Note the mandibular osteotomies and internal distraction hardware.
-
Left-lateral cranial radiograph showing mandibular osteotomy and distraction osteogeneses for micrognathia.
-
Lateral radiograph of same patient as in Media files 7 and 8. Completion of distraction; note increased mandibular length.




