Background
Malabsorption syndromes encompass numerous clinical entities that result in chronic diarrhea, abdominal distention, and failure to thrive. [1] Clinical malabsorption can be broken down into several distinct conditions, both congenital and acquired, that affect one or more of the different steps in the intestinal hydrolysis and subsequent transport of nutrients.
The major site of absorption is the small intestine, as depicted in the illustration below.
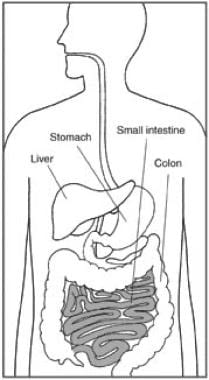 The small intestine is a major site of absorption.
The small intestine is a major site of absorption.
Pathophysiology
Carbohydrate, fat, or protein malabsorption is caused by a disorder in the intestinal processes of digestion, transport, or both of these nutrients across the intestinal mucosa into the systemic circulation. Either a congenital abnormality in the digestive or absorptive processes or, more commonly, a secondarily acquired disorder of such processes may result in malabsorption.
Carbohydrates
Of the carbohydrates most commonly present in the diet (starches, sucrose, lactose), only starches require preliminary luminal digestion by salivary and, more importantly, pancreatic amylases. Despite the slow development of pancreatic amylase, whose secretion reaches adult levels during the end of the first year of life, cooked starch malabsorption is rare in infants because of the activity of the brush-border bound glucoamylase, an esoglycosidase that develops early in life.
The final products of amylase digestion include maltose, maltotriose, and higher residues of glucose polymers. The final hydrolysis of disaccharides and oligosaccharides occurs at the brush border of the enterocytes, where sucrase-isomaltase breaks down maltose, isomaltose (to glucose), and sucrose (to glucose and fructose); glucoamylase releases glucose from glucose polymers; and lactase splits lactose into glucose and galactose. Subsequent entry of the final monosaccharides (glucose, galactose, fructose) into the enterocytes through the brush border occurs via carrier molecules. Glucose and galactose share the same carrier, SGLT-1, which transports one molecule of the monosaccharide and one molecule of sodium (Na) in a secondarily active transport, energized by Na-activated and potassium (k)-activated adenosine triphosphatase (NaK ATPase). Instead, fructose is transported by Glut2 and Glut5 transporters across the cell membrane. Although Glut2 can transport both glucose and fructose, Glut5 is a fructose-specific transporter, working only down a concentration gradient (facilitated diffusion).
Disorders of these processes can be congenital (cystic fibrosis and Shwachman-Diamond syndrome, which may cause amylase deficiency; the extremely rare congenital lactase deficiency; glucose-galactose malabsorption; sucrase-isomaltase deficiency; adult-type hypolactasia) or acquired: the most common being lactose intolerance, typically secondary to a damage of the mucosa, such as a viral enteritis or conditions that cause mucosal atrophy, such as celiac disease.
Protein
Proteins are first digested in the stomach, where pepsinogens, which are activated to pepsins by a pH of less than 4, hydrolyze them in large molecular weight peptides. Upon entering the duodenum, the pancreatic proteases (activated by trypsin, secreted by the pancreas as a proenzyme, trypsinogen, which is subsequently activated by the brush border–bound enterokinase) further split them into low molecular weight peptides and free amino acids.
Interestingly, the final breakdown products of intraluminal digestion of protein are composed of low molecular weight peptides (2-6 amino acid residues) for 70% and of free amino acids for 30%. Subsequently, brush border–bound peptidases further hydrolyze peptides to release a mixture of free amino acids and small peptides (2-3 amino acid residues). Finally, free amino acids are taken up by enterocytes through specific Na-linked carrier systems (5 carriers with selective affinities for groups of amino acids are described), whereas dipeptides and tripeptides are translocated into the absorptive epithelial cells by the peptide transporter 1 (PEPT1), which is a carrier with a broad specificity linked to H entry. [2] In the first few months of life, the latter system is much more active than those that transport amino acids and is thought to play a bigger physiological role.
Congenital disorders of protein digestion include conditions such as cystic fibrosis, Shwachman-Diamond syndrome, and enterokinase deficiency, which cause inadequate intraluminal digestion. No congenital defects have been described in any of the brush border–bound peptidases or in the peptide carrier.
Acquired disorders of protein digestion and/or absorption are nonspecific (ie, they also affect the absorption of carbohydrates and lipids) and are found in conditions that result in damage to the absorptive intestinal surface, such as extensive viral enteritis, milk protein allergy enteropathy, and celiac disease.
Lipids
A lingual lipase is responsible for the first partial hydrolysis of triglycerides; this enzyme becomes active in persons with low gastric pH levels and is active even in premature infants. However, the largest part of triglyceride digestion is accomplished in the duodenojejunal lumen because of a complex of pancreatic enzymes, the most important of which is the lipase-colipase complex. Like amylase, these enzymes also develop slowly, and this accounts for the known low capacity of babies to absorb lipids, termed physiologic steatorrhea of the newborn. Additionally, adequate concentrations of intraluminal conjugated bile salts are needed to form micelles, and the secretion of bile acids may also be partially inadequate in very young patients.
Disorders of these processes can be congenital (cystic fibrosis and Shwachman-Diamond syndrome, which cause lipase and colipase deficiency; the uncommon isolated deficiency of lipase and colipase; the extremely rare congenital primary bile acid malabsorption, which results in low bile acids concentrations) or acquired (secondary mostly to disorders of the liver and the biliary tract or to chronic pancreatitis). Clearly, any condition that results in the loss of small intestinal absorptive surface also causes steatorrhea.
Epidemiology
Frequency
Genetically determined syndromes
The prevalence of celiac disease in the United States is around 1% of the general population [3] and has increased over time. [4] Celiac disease in its entirety (ie, including the forms without overt malabsorption) is by far the most common inherited malabsorption syndrome. Cystic fibrosis is the second most common malabsorption syndrome. Other congenital disorders are rare, with the exception of adult-type hypolactasia, which has a prevalence that varies greatly among different ethnic groups.
Acquired syndromes
Cow's milk and soy milk protein allergies are common, especially in infants and young children. [5] The prevalence of milk protein allergy, of which enteropathy is one of the presenting clinical symptoms, is estimated to be around 3%. A transient and common form of malabsorption in infants results from acute-onset enteritis (mostly viral, specifically rotaviral), which causes transient lactose intolerance. Although toddler's (or unspecific) diarrhea accounts for approximately 7.5% of referrals to pediatric gastroenterologists, it should not be considered a malabsorption syndrome because, by definition, no digestive or absorptive processes are impaired.
Secondary malabsorption syndromes that result from liver, pancreas, and intestinal diseases are uncommon. The manifestations vary according to the severity of each disease and the extent of intestinal mucosal injury.
Mortality/Morbidity
Although the morbidity can be severe, and aside from the single entity of cystic fibrosis, common malabsorption syndromes carry low mortality rates. Neonates and young infants, especially those with signs of malnutrition, are particularly at risk. In many of the congenital syndromes, morbidity varies with the particular syndrome and may be associated with systemic manifestations of the disease.
Race
Congenital sucrase-isomaltase deficiency is most common in Canadian Eskimos and natives of Greenland. Deficiency of trehalose, a sugar found almost exclusively in mushrooms, is rare, except in natives of Greenland. Adult-onset lactase deficiency is most common in persons of Asian, African, and Mediterranean descent.
Sex
Celiac disease is slightly more common in females. Autoimmune enteropathy is an X-linked disorder that only affects males in familial cases.
Age
Neonates and young infants with malabsorption syndromes are at particularly high risk for chronic diarrhea and malnutrition. Symptoms of a congenital disease are usually apparent shortly after birth (the exception being adult-onset lactase deficiency, which appears only after age 6-8 y) or after a short hiatus once a particular substance is ingested in substantial amounts. Protein sensitivity syndromes to milk or soy protein usually present in infants younger than 3 months, but solid food protein sensitivity syndromes are also known to occur in older patients.
-
The small intestine is a major site of absorption.