Practice Essentials
Lymphomas are malignant neoplasms of lymphoid lineage. Broadly classified as either Hodgkin disease (Hodgkin's disease) or as non-Hodgkin lymphoma, lymphomas are clinically, pathologically, and biologically distinct (see the image below). [1, 2] (See Prognosis and Workup.)
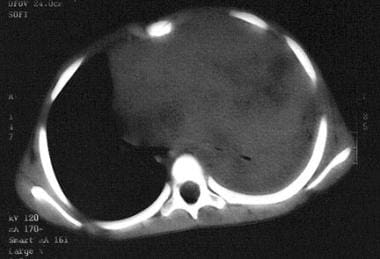 Massive mediastinal T-lymphoblastic lymphoma. Note compression of the left mainstem bronchus and the pulmonary atelectasis.
Massive mediastinal T-lymphoblastic lymphoma. Note compression of the left mainstem bronchus and the pulmonary atelectasis.
According to the National Cancer Institute (NCI) formulation, most childhood non-Hodgkin lymphomas can be classified as one of the following types:
-
Lymphoblastic lymphomas
-
Small noncleaved cell lymphomas (SNCCLs) - Burkitt lymphomas and non-Burkitt lymphomas (Burkittlike lymphomas)
-
Large cell lymphomas (LCLs)
B-cell LCLs and anaplastic (usually T-cell) LCLs (ie, Ki-1+ lymphomas) have come to be viewed as distinct entities. In this article, these categories are considered separately. Other, less common forms of childhood lymphoma (some of which are much more common in adults) are not discussed.
Disease progression
Most malignancies arise as disease localized in the organ or tissue of origin. They may then secondarily spread by means of local extension or distant metastases. In contrast, non-Hodgkin lymphoma is best regarded as a systemic disease, because of the unique anatomy of the lymphoid system and because of the physiology of lymphoid cells, which tend to migrate whether they are normal or malignant. The role of lymphoma stem cells in the genesis and maintenance of B-cell lymphomas remains speculative. [3] (See Etiology.)
Non-Hodgkin lymphoma versus acute leukemia
Childhood non-Hodgkin lymphoma generally manifests as bulky extramedullary (usually extranodal) disease with or without demonstrable dissemination. Particularly in the case of lymphoblastic diseases, the distinction between non-Hodgkin lymphoma and acute leukemia is arbitrary. Therefore, these entities (acute lymphoblastic leukemia and lymphoblastic lymphoma) are best considered in terms of a spectrum ranging from clinically localized disease to overt leukemia.
In most treatment protocols, acute leukemia is now defined on the basis of marrow involvement above some threshold (typically, a blast count of >25%) irrespective of the presence of bulky extramedullary disease. In contrast, an extramedullary tumor accompanied by marrow involvement below this threshold constitutes stage 4 lymphoma. The term, "leukemia/lymphoma syndrome," which was once in common usage, is no longer valid.
Management
Since the late 1960s, treatment outcomes for pediatric patients with non-Hodgkin lymphoma have steadily improved. Even for patients with advanced disease, event-free survival rates are now 65-90%. (See Prognosis, Treatment, and Medication.)
The mainstay of conventional therapy is multiagent chemotherapy tailored to the histologic subtype and the clinical stage of disease. In certain individuals with non-Hodgkin lymphoma, surgical resection and radiation therapy are also key components of definitive treatment. Newer therapies that target immunologic and biologic aspects of the lymphoma are still under development but beginning to appear in the clinical arena. (See Treatment and Medication.)
Patient education
For patient education information, see the Cancer Center, as well as Lymphoma (Hodgkin’s Disease and Non-Hodgkin’s Lymphoma) and Cancer of the Mouth and Throat.
Etiology
In industrialized countries, most individuals with non-Hodgkin lymphoma have no known etiology or association. Epidemiologic data suggest that certain human leukocyte antigen (HLA) types, and even certain blood types, may increase or decrease the likelihood of developing non-Hodgkin lymphoma. [4, 5]
Findings from several epidemiologic studies suggest that pesticide exposure may play a role in the development of adult non-Hodgkin lymphoma; the case for its involvement in childhood non-Hodgkin lymphoma is less compelling than the case for adults, but this is still under investigation. [6, 7]
Exposure to air pollution may increase the risk of pediatric non-Hodgkin lymphoma. Hvidtfeldt et al observed an association between residential exposure to fine atmospheric particulate matter, including black carbon, and the development of non-Hodgkin lymphoma in children. [8] Another study by the same team of Danish researchers confirmed their earlier findings and also suggested a role for exposure to secondary inorganic aerosols. [9]
The epidemiologic association between non-Hodgkin lymphoma and certain paternal occupations (eg, those that increase contact with other individuals) suggests a possible infective etiology for childhood non-Hodgkin lymphoma. [10]
An interesting statistical association exists between high birth weight and the subsequent risk of childhood cancers, including non-Hodgkin lymphoma. [11]
Regarding protective factors, results of one case-control study suggested that exposure to sunlight may protect individuals against non-Hodgkin lymphoma, presumably because of enhanced vitamin D synthesis. [12]
Immunosuppression and viral infection
Immunosuppressed individuals, such as those with human immunodeficiency virus (HIV) infection or those who have undergone solid organ transplantation or bone marrow transplantation, are at increased risk for developing non-Hodgkin lymphoma, particularly small noncleaved cell lymphomas (SNCCL) and large cell lymphomas (LCLs) of B-cell origin. The Epstein-Barr virus, which causes B-cell proliferation and in vitro immortalization, has been implicated in most of these lymphomas. Primary central nervous system (CNS) lymphoma is more common in these patients than in others.
Previous malignancy
Patients successfully treated for Hodgkin disease are at increased risk for developing non-Hodgkin lymphoma. This phenomenon appears to reflect the combined effects of chemotherapy and radiotherapy, as well as the immunosuppressive effects of Hodgkin disease. Adults older than 40 years who received combined-modality therapy are at particular risk; their 15-year incidence of non-Hodgkin lymphoma is as high as 39%. [13]
Splenectomy, now rarely performed in patients with Hodgkin disease, is another reported risk factor for secondary malignancies, including non-Hodgkin lymphoma. [14]
Secondary non-Hodgkin lymphoma is less common among pediatric patients who survive cancer than among adults. A cohort of 5484 children was treated for various malignancies at St Jude Children's Research Hospital. Over 30,710 person-years of follow-up care, only 3 had secondary non-Hodgkin lymphoma. The 15-year actuarial risk of non-Hodgkin lymphoma was 0.16% in this group.
However, even among children, patients treated for Hodgkin disease are particularly at risk. A literature review revealed 24 incidents of secondary non-Hodgkin lymphoma among patients whose primary malignancy had been diagnosed when they were younger than 20 years. Eighteen (75%) of the patients previously had Hodgkin disease. [15]
Geographic location
In sub-Saharan Africa, the development of endemic Burkitt lymphoma is strongly associated with previous exposures to malaria (with resultant T-cell suppression) and the Epstein-Barr virus. Speculation suggests that mosquito-borne arboviruses may also play a role in the development of Burkitt lymphoma in this part of the world.
In addition, exposure to 4-deoxyphorbol ester from the plant Euphorbia tirucalli (by means of goat's milk) is tentatively implicated in the pathogenesis of endemic Burkitt lymphoma. [16, 17]
Genetics
The genetic basis of pediatric non-Hodgkin lymphoma has been studied extensively. [18] Each subtype of non-Hodgkin lymphoma is characterized by 1 or more molecular alterations that contribute to the malignant phenotype. Many of these alterations are chromosomal translocations involving genes for immunoglobulin or T-cell receptor (TCR) molecules.
During normal lymphocyte development, these loci undergo recombination that enhances immunologic diversification. However, mistargeted recombination leads to translocations with other genes, typically those that regulate cell growth. The resulting dysregulation of these other genes contributes to the transformed phenotype.
For example, the hallmark of Burkitt lymphoma is a t(8;14)(q24;q32) translocation, which is observed in approximately 80% of patients. This translocation juxtaposes c-myc, which encodes a transcription factor important in initiation of the cell cycle, with the locus for the immunoglobulin heavy chain. Less commonly, c-myc is adjoined to the gene encoding the immunoglobulin kappa light chain [t(2;8)(p11;q24)] or the lambda light chain [t(8;22)(q24;q11)].
In all 3 instances, the result is aberrant expression of the c-MYC protein under the influence of regulatory sequences of immunoglobulin genes. This aberration contributes to the pathogenesis of Burkitt lymphoma. [19]
Aside from the t(8;14) translocation, Burkitt lymphoma frequently involves a gain of chromosomal material that can affect any of a number of chromosomes. Abnormalities of chromosomal arms 1q, 7q, or 13q may portend a poor prognosis. [19, 20]
A small portion of T-lymphoblastic lymphomas are also associated with translocations involving one of the TCR loci; either TCR alpha delta (14q11) or TCR beta (7q34). The most common example (observed in 7% of children with T-lymphoblastic lymphomas) is the t(11;14)(p13;q11) translocation, which enhances expression of the LMO2 gene on chromosome 11. This gene encodes LIM protein, an apparent modulator of gene transcription.
A more common abnormality observed in T-lymphoblastic lymphoma is a deletion in a regulatory region of the gene TAL1. This deletion, which is too small to be detected with conventional cytogenetic techniques, leads to aberrant expression of Tal-1, another transcriptional regulator.
Inactivation of the multiple tumor-suppressor gene 1 (MTS-1/p16INK4 alpha/CDKN2) on chromosome 16 has been identified as a common genetic event in T-cell ALL; its frequency in T-lymphoblastic lymphoma is likely to be a significant factor. Of interest, the deletions or disruptions responsible for this inactivation are apparently related to illegitimate activity of the same V(D)J recombinase that mediates recombination of the TCR gene. [21] Thus, even in the absence of a TCR translocation, similar molecular mechanisms may be responsible for disrupting other genes involved in normal control of the cell cycle.
Some B-lineage LCLs have the same t(8;14)(q24;q32) translocation observed in Burkitt lymphoma. Compared with adults with B-LCL, this appears to be more common in children and may portend a worse prognosis. [22] Alternatively, most anaplastic (T-lineage) LCLs in children involve a t(2;5)(p23;q35) translocation. This change joins the nucleophosmin gene (NPM) on chromosome 5 to a gene called anaplastic lymphoma kinase (ALK) on chromosome 2 and allows for the expression of p80, an NPM/ALK fusion protein.
Rarely, anaplastic LCLs exhibit rearrangements of c-myc; based on small numbers of patients, this appears to confer a poor prognosis. [23, 24]
Transcripts of NPM/ALK are also observed in about 20% of individuals with non-Hodgkin lymphoma lacking cytogenetic evidence of t(2;5); this finding reflects an occult or variant translocation. [25] Patients with non-Hodgkin lymphomas expressing p80 may have a survival advantage over patients whose lymphomas lack p80. [26]
Epidemiology
United States statistics
Taken collectively, lymphomas are the third most common childhood malignancies after acute leukemias (acute lymphoblastic leukemia, acute myelocytic leukemia) and brain tumors, [27] constituting 10-12% of childhood cancers. In older adolescents, lymphomas surpass brain tumors in incidence, largely because of the increased frequency of Hodgkin disease in this age group.
National cancer data from the NCI Surveillance, Epidemiology, and End Results (SEER) program for 2002-2006 are shown below. In children, non-Hodgkin lymphoma is somewhat less common than Hodgkin disease. However, the incidence of non-Hodgkin lymphoma appears to be rising in the United States. This trend largely reflects the occurrence of non-Hodgkin lymphoma in patients who are immunocompromised (eg, patients with HIV) and in patients who were previously exposed to chemotherapy and irradiation as treatment for an unrelated cancer.
Age-adjusted incidences of selected cancers per 100,000 individuals aged 0-19 years are as follows [27] :
-
All sites - 16.6
-
Leukemias - 4.5
-
Brain and other nervous tissues - 2.9
-
Hodgkin disease - 1.2
-
Non-Hodgkin lymphoma - 1.1
-
Soft tissue - 1.1
-
Bone and joint - 0.9
-
Kidney and renal pelvis - 0.6
International statistics
A study reviewing non-Hodgkin lymphoma rates in Canada between 1970 and 1996 found that the incidence of non-Hodgkin lymphoma apparently increased in that nation over the 3 decades of the report. [28] The cause for this rise is unclear.
Burkitt lymphoma is significantly more common in sub-Saharan Africa than in other parts of the world, accounting for approximately one half of all childhood cancers in that region. The disease’s incidence also appears to be higher in Latin America, North Africa, and the Middle East than it is in the United States or Europe.
A retrospective study of pediatric lymphoma in a hospital in Pakistan determined that non-Hodgkin lymphoma accounted for 75% of these cases.
Race- and sex-related demographics
In the United States, the incidence of non-Hodgkin lymphoma is twice as high among whites as it is among blacks, with respective rates of 9.1 and 4.6 cases per million individuals per year.
In addition, the incidence of non-Hodgkin lymphoma in the United States is almost twice as high in males as in females. For 2002-2006, the SEER age-adjusted incidence of non-Hodgkin lymphoma was 1.4 cases per 100,000 males (age 0-19 years) and 0.8 cases per 100,000 females. [27]
Age-related demographics
In the United States, the age-specific incidence of non-Hodgkin lymphoma only slightly increases over the first 2 decades of life. By comparison, the incidence of Hodgkin disease increases more dramatically as children age. In adulthood, the risk of non-Hodgkin lymphoma steadily climbs, whereas the age-specific incidence of Hodgkin disease is biphasic.
A study by Mbulaiteye et al of 3,403 cases of Burkitt lymphoma spread over 4 continents found that in all regions and over all periods in the study (which covered 1963-2002), peaks in the rate of Burkitt lymphoma occurred close to the ages of 10 and 70 years. The investigators concluded that their findings—which also included other age-related differences in the disease rate, as well as age-related differences in the male-to-female ratio for the disease’s occurrence—supported their hypothesis that Burkitt lymphoma is multimodal and that the age-related peaks in the disease may aid in determining differences in the disorder’s etiology and/or biology at the peak ages. [29]
Prognosis
The overall prognosis for children with non-Hodgkin lymphoma has steadily improved. Period analysis of SEER data for children under 15 years showed that 5- and 10-year survival increased, respectively, from 76.6% and 73.0% in 1990-1994 to 87.7% and 86.9% in 2000-2004. The projected 10-year survival rate for children diagnosed in 2005-2009 was 90.6%. [30]
Among patients with non-Hodgkin lymphoma, the major determinants of prognosis are histology and disease stage. The presence or absence of particular molecular markers (eg, anaplastic lymphoma kinase (ALK) and/or CD56 in anaplastic large cell lymphoma [LCL]) has additional prognostic significance. [31]
Age at diagnosis is a significant prognostic factor when one considers the older pediatric patient (adolescent or young adult) with non-Hodgkin lymphoma. Broadly speaking, older patients have poorer outcomes, [32] and there is increasing recognition that these patients need to be viewed as a unique population in terms of disease biology and treatment tolerance. [33]
Studies have also defined host (ie, nontumoral) prognostic factors for patients with non-Hodgkin lymphoma. For example, polymorphisms of immune-related genes, such as those for interleukin (IL)–10 and tumor necrosis factor, show significant associations with treatment outcomes in adults with non-Hodgkin lymphoma. [34, 35] Similar pediatric data are not yet available.
CNS lymphoma
Pediatric and adolescent patients with CNS lymphoma have better outcomes than do adults with this disease. [36] An Eastern Cooperative Oncology Group (ECOG) performance status score of 0-1 is associated with improved survival. Higher dose methotrexate is associated with slightly better response.
Relapsed or refractory disease
Patients with relapsed or refractory non-Hodgkin lymphoma are candidates for salvage therapy, which often includes autologous or allogeneic hematopoietic stem cell transplantation. The likelihood of cure depends on diagnosis, initial therapy, and length of first remission. [37] Even patients who experience relapse after autologous transplantation are potentially salvageable with a second transplant procedure. [38]
Complications
Growth
Linear growth of the pediatric patient often slows during aggressive chemotherapy. Most patients, however, have catch-up growth and eventually achieve a height in the normal range. Clinically significant, long-term growth retardation is essentially confined to patients who receive cranial irradiation. Of interest, a notable minority of children treated for lymphoma eventually becomes obese; the basis for this effect is unclear. [39]
Neuropsychological sequelae
Neurotoxicity due to combined cranial irradiation and intrathecal chemotherapy is well described in patients with acute lymphoblastic leukemia (ALL). Neurotoxic effects range from mild learning disabilities to a profound necrotizing leukoencephalopathy. Patients with CNS lymphoma are at risk for developing these same complications.
In the absence of radiation, intrathecal chemotherapy has been thought have little effect on neuropsychological function. Data now suggest, however, that patients with non-Hodgkin lymphoma who survive without irradiation are more likely to require special education classes than are their siblings.
In a recent report from Finland, scholastic achievement was particularly impaired in survivors of childhood non-Hodgkin lymphoma and not in patients with Wilms tumor or Hodgkin disease. [40]
The peripheral neuropathy associated with vincristine occasionally leaves permanent deficits, particularly lower extremity weakness.
Fertility
Alkylating agents have particularly been implicated in acute gonadal dysfunction. The long-term effects of these agents among survivors of childhood cancer are somewhat unclear.
Prepubertal boys appear to be at low risk for eventually developing azoospermia or failure of sexual maturation. Older male adolescents are at some risk for developing temporary azoospermia; they can perhaps consider banking their semen before undergoing chemotherapy, if this is feasible.
Ovarian failure after high-dose alkylator therapy has also been described. Nonetheless, a report found that female survivors of non-Hodgkin lymphoma had little or no apparent deficit in pregnancies.
Patients who have a relapse, particularly those treated with myeloablative chemotherapy and/or total body irradiation, have a particularly elevated risk of developing permanent gonadal dysfunction.
Secondary malignancies
The oncogenic potential of therapeutic radiation is well documented, but the risk of secondary malignancies associated with chemotherapy is less obvious. One clearly implicated antineoplastic agent is etoposide. However, the risk of secondary acute myelocytic leukemia (AML) due to this drug appears to be insignificant at cumulative doses of less than 1000 mg/m2.
Cyclophosphamide has also been identified as a potential carcinogen. The relative risk of secondary malignancies in children exposed to cyclophosphamide is estimated to be as high as 7.4 if the cumulative exposure is more than 13 g/m2.
In one series, however, only 2 cases of secondary cancer (1 case of malignant melanoma and 1 case of spindle-cell sarcoma, which arose in a radiation field) were found among 86 survivors of pediatric non-Hodgkin lymphoma. The 86 patients were evaluated for a mean period of 11 years after diagnosis. [41] These findings suggest that, despite concerns about the effects of chemotherapy, patients who do not receive irradiation are unlikely to develop a secondary malignancy. However, even longer follow-up is needed to accurately assess the lifelong risk of secondary malignancies.
Fortunately, the risk of secondary cancer appears to be decreasing, due to the recognition (and relative avoidance) of treatment-related risk factors such as radiation and high-dose epipodophyllotoxins. [42]
Cardiotoxicity
At high cumulative doses, doxorubicin is likely to cause delayed myocardial toxicity. [43] Irradiation of the heart exacerbates this effect.
In a recent report, 7 of 29 survivors (aged 2-39 years at diagnosis) who received doxorubicin 240-560 mg/m2 eventually developed left ventricular dysfunction approximately 10 years later. However, other reports have described anthracycline-related cardiotoxicity after cumulative doses as small as 100 mg/m2.
If patients have received more than 300 mg/m2 of doxorubicin, perform screening echocardiography every 2-4 years on an indefinite basis. Lower this threshold if mediastinal irradiation was also administered.
Skeletal toxicity
Long-term, high-dose steroid therapy is associated with osteoporosis and avascular necrosis of bone. In one report, long-term survivors of acute lymphoblastic leukemia and non-Hodgkin lymphoma exhibited low bone mineral density in roughly two thirds of men and one third of women. The effects of dexamethasone therapy, cranial radiation, and bone marrow transplantation appeared to be additive. [44]
Avascular necrosis most commonly affects the femoral heads, and it may be associated with a slipped capital femoral epiphysis. Avascular necrosis of bone is most often observed in adolescents and in female patients. The spectrum of disease ranges from asymptomatic radiographic findings to incapacitating joint destruction requiring restorative surgery.
Radiation therapy is associated with osteopenia. This may occur locally or, of interest, it may be observed diffusely after cranial irradiation. [45]
Viral transmission by means of blood products
Transmission of cytomegalovirus (CMV) is possible if unscreened blood products are administered without prior leukoreduction. Since leukoreduction reduces the risk of CMV transmission, the role for CMV-seronegative blood products is controversial; arguably, these products should be given to patients who may eventually undergo bone marrow transplantation (BMT), but the benefits of this practice are marginal.
With modern transfusion practices, exposure to hepatitis B or C virus is rare. Nonetheless, patients occasionally demonstrate serologic evidence of exposure. Chronic active hepatitis and hepatocellular carcinoma are potential sequelae of this exposure.
Explain the risks of viral transmission to patients, their parents, and/or caregivers before transfusions are given.
Mediastinal involvement
Individuals with lymphoblastic lymphoma often present with mediastinal involvement, which may be massive and life threatening. Airway compression is a particular concern and must be considered in any patient with neck or chest disease. (See the image below.)
Massive mediastinal T-lymphoblastic lymphoma. Note compression of the left mainstem bronchus and the pulmonary atelectasis.
Even in the absence of symptomatic airway compromise, sudden obstruction may be a risk if the patient undergoes anesthesia for biopsy or placement of a central line. In these individuals, consider biopsy performed under local anesthesia or immediate radiation therapy to the airway, provided that another site of disease is outside the radiation field (to allow for subsequent histologic confirmation of the diagnosis).
Mediastinal tumors may cause compression of the great vessels (superior vena cava syndrome), with swelling of the neck, face, and upper extremities. Esophageal compression may lead to dysphagia. Pleural effusion is sometimes observed and may be large enough to cause symptoms. In affected individuals, thoracentesis may be therapeutic and diagnostic, obviating biopsy. (See the image below.)
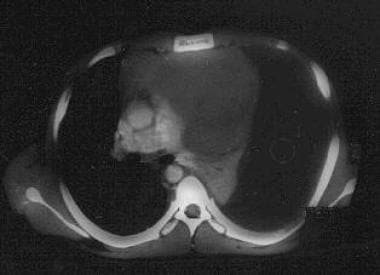 Massive left pleural effusion as a complication of an upper anterior mediastinal T-lymphoblastic lymphoma. Note the atelectatic left lung. The diagnosis was established by means of thoracentesis. This patient had presented with bilateral parotid gland enlargement.
Massive left pleural effusion as a complication of an upper anterior mediastinal T-lymphoblastic lymphoma. Note the atelectatic left lung. The diagnosis was established by means of thoracentesis. This patient had presented with bilateral parotid gland enlargement.
Bowel obstruction
In the United States, most patients with small noncleaved cell lymphomas (SNCCLs) present with abdominal involvement, typically in the ileocecal area and arising from Peyer patches. A potential complication at the time of diagnosis is bowel obstruction due to direct compression, torsion, or intussusception. Because of bowel perforation, some patients have ascites or present with a clinical picture of acute appendicitis or peritonitis. (See the images below.)
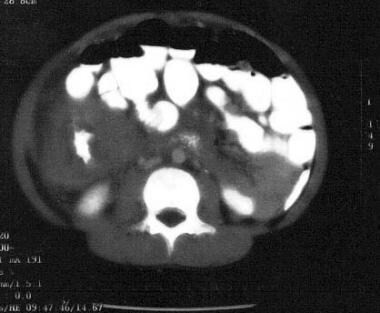 Non-Hodgkin lymphoma of the terminal ileum. Note the doughnut sign, ie, intraluminal contrast material surrounded by a grossly thickened bowel wall. This appearance is highly suggestive of small noncleaved cell lymphoma (Burkitt type).
Non-Hodgkin lymphoma of the terminal ileum. Note the doughnut sign, ie, intraluminal contrast material surrounded by a grossly thickened bowel wall. This appearance is highly suggestive of small noncleaved cell lymphoma (Burkitt type).
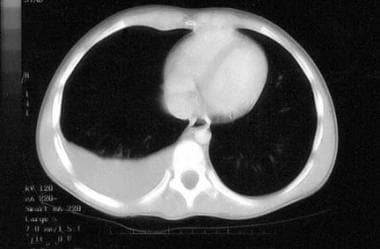 Malignant pleural effusion. Non-Hodgkin lymphoma of the terminal ileum was diagnosed; the doughnut sign (ie, intraluminal contrast material surrounded by a grossly thickened bowel wall) was present. A diagnosis of stage 3 Burkitt lymphoma was established by means of pleurocentesis. (The bone marrow was normal.) The patient was treated successfully and never required an abdominal procedure.
Malignant pleural effusion. Non-Hodgkin lymphoma of the terminal ileum was diagnosed; the doughnut sign (ie, intraluminal contrast material surrounded by a grossly thickened bowel wall) was present. A diagnosis of stage 3 Burkitt lymphoma was established by means of pleurocentesis. (The bone marrow was normal.) The patient was treated successfully and never required an abdominal procedure.
In equatorial Africa, SNCCL (ie, endemic Burkitt lymphoma) classically appears as a mass in the jaw, nasopharynx, or orbit. These masses grow rapidly and can be disfiguring.
Other complications
Rapidly growing or bulky tumors can cause severe metabolic derangement, which may be life threatening. One indicator of the potential for tumor lysis syndrome is an elevated plasma lactate dehydrogenase level or hyperuricemia at the time of diagnosis. The start of effective chemotherapy acutely increases the risk of complications, including hyperkalemia, hyperphosphatemia, hypocalcemia, oliguria, and renal failure.
Other immediate risks depend on the site and extent of involvement. These in turn vary according to the histologic subtype of disease.
With current treatments, non-Hodgkin lymphomas in most children are apparently curable. The results depend on achieving a precise histologic diagnosis, thorough staging of the disease, and applying complex, multiagent (and sometimes multimodal) treatment. The short-term morbidity of chemotherapy regimens is considerable, but the effects are usually manageable. Late effects of treatment are a growing concern, as survival rates are increasing.
-
Massive mediastinal T-lymphoblastic lymphoma. Note compression of the left mainstem bronchus and the pulmonary atelectasis.
-
Non-Hodgkin lymphoma of the terminal ileum. Note the doughnut sign, ie, intraluminal contrast material surrounded by a grossly thickened bowel wall. This appearance is highly suggestive of small noncleaved cell lymphoma (Burkitt type).
-
Malignant pleural effusion. Non-Hodgkin lymphoma of the terminal ileum was diagnosed; the doughnut sign (ie, intraluminal contrast material surrounded by a grossly thickened bowel wall) was present. A diagnosis of stage 3 Burkitt lymphoma was established by means of pleurocentesis. (The bone marrow was normal.) The patient was treated successfully and never required an abdominal procedure.
-
Massive left pleural effusion as a complication of an upper anterior mediastinal T-lymphoblastic lymphoma. Note the atelectatic left lung. The diagnosis was established by means of thoracentesis. This patient had presented with bilateral parotid gland enlargement.
Tables
- Table 1. Modified LSA2 L2 Therapy in Children's Cancer Group Protocol 552
- Table 2. Therapy for Stage III and IV Non–B-Cell Disease* According to BFM Protocol 86
- Table 3. Clinical Risk Groups in the International Trial for Patients With SNCCL (Children's Cancer Group study 5961)
- Table 4. Standard Therapy in the International Trial for Patients With SNCCL, Group A*
- Table 5. Standard Therapy in the International Trial for Patients With SNCCL, Group B*
- Table 6. Standard Therapy in the International Trial for Patients With SNCCL, Group C*
- Table 7. Prephase Therapy for Ki-1+ Anaplastic LCLs According to the ALCL99 Protocol
- Table 8. Subsequent Therapy for Ki-1+ Anaplastic LCLs According to the ALCL99 Protocol (alternating cycles, repeated 3times each)
Phase |
Drug |
Route |
|
Induction |
Cyclophosphamide, vincristine, daunorubicin |
IV |
|
Ara-C, methotrexate |
IT |
||
Prednisone |
PO |
||
Consolidation |
Ara-C |
IV or SC |
|
6-thioguanine |
PO |
||
Methotrexate |
IT |
||
L-asparaginase |
IM |
||
BCNU |
IV |
||
Phase |
Cycle |
Drug |
Route |
Maintenance* |
1 |
6-thioguanine |
PO |
Cyclophosphamide |
IV |
||
2 |
Hydroxyurea |
PO |
|
Daunorubicin |
IV |
||
3 |
Methotrexate |
PO |
|
BCNU |
IV |
||
4 |
Ara-C |
IV or SC |
|
Vincristine |
IV |
||
Source: Children's Cancer Group Ara-C = cytarabine; BCNU = 1,3-bis(2-chloroethyl)-1-nitrosourea, or carmustine; IM = intramuscular; IT = intrathecal; IV = intravenous; PO = oral; SC = subcutaneous * A minimum of 5 repeated courses (total duration of therapy >18 mo) are noted. Each course of intrathecal methotrexate (day 0 of each course) consists of 4 cycles of rotating drug pairs that are administered every 2 weeks after blood counts have recovered. |
|||
Phases |
Drug |
Route |
Induction |
Prednisone, 6-mercaptopurine |
PO |
Vincristine, daunorubicin, cyclophosphamide, Ara-C |
IV |
|
L-asparaginase |
IM |
|
Methotrexate |
IT |
|
Consolidation |
6-mercaptopurine |
PO |
Methotrexate with leucovorin rescue |
IV |
|
Methotrexate |
IT |
|
Re-induction |
Dexamethasone, 6-thioguanine |
PO |
Vincristine, doxorubicin, cyclophosphamide, Ara-C |
IV |
|
L-asparaginase |
IM |
|
Methotrexate |
IT |
|
Maintenance† |
6-mercaptopurine, methotrexate |
PO |
Source: Berlin-Frankfurt-Munster Group Ara-C = cytarabine; IT = intrathecal; IV = intravenous; PO = oral; SC = subcutaneous * Diagnoses included lymphoblastic lymphoma of the T-cell or precursor B-cell type, immunoblastic T-cell lymphoma, and other peripheral T-cell lymphomas. Of note, patients with Ki-1+ anaplastic large cell lymphomas (LCLs) were not included. † Continued until 24 months after diagnosis. |
||
Clinical Group |
Subjects, Estimated % |
Definition |
A |
10 |
All resected stage I or abdominal stage II tumors |
B |
65 |
Unresected stage I or II tumor, stage III, tumor, or stage IV tumor with no CNS involvement and < 25% marrow blasts |
C |
25 |
CNS involvement or >25% marrow blasts |
Drug |
Route |
Prednisone |
PO |
Vincristine, cyclophosphamide, doxorubicin |
IV |
Filgrastim (G-CSF), to enhance neutrophil recovery |
SC or IV |
G-CSF = granulocyte colony-stimulating factor; IV = intravenous; PO = oral; SC = subcutaneous * See Table 3 for the definition of group A. All subjects received 2 cycles. |
|
Phase |
Drug |
Route |
|
Reduction |
Prednisone |
PO |
|
|
Vincristine, cyclophosphamide |
IV |
|
|
Methotrexate/hydrocortisone |
IT |
|
Phase |
Cycles |
Drug |
Route |
Induction |
2, starting 7 days after reduction |
Prednisone |
PO |
Vincristine, methotrexate with leucovorin rescue, cyclophosphamide, doxorubicin |
IV |
||
Methotrexate/hydrocortisone |
IT |
||
Filgrastim (G-CSF) |
SC or IV |
||
| Rituximab | IV | ||
Consolidation |
2 |
Methotrexate with leucovorin rescue, Ara-C |
IV |
Methotrexate/hydrocortisone, Ara-C/hydrocortisone |
IT |
||
Filgrastim (G-CSF) |
SC or IV |
||
| Rituximab | IV | ||
Maintenance** |
1 |
Prednisone |
PO |
Vincristine, methotrexate with leucovorin rescue, cyclophosphamide, doxorubicin |
IV |
||
Methotrexate/hydrocortisone |
IT |
||
Ara-C = cytarabine; G-CSF = granulocyte colony-stimulating factor; IT = intrathecal; IV = intravenous; PO = oral, SC = subcutaneous * See Table 3 for the definition of group B. |
|||
Phase |
Drug |
Route |
|
Reduction |
Prednisone |
PO |
|
Vincristine, cyclophosphamide |
IV |
||
Methotrexate/Ara-C/hydrocortisone |
IT |
||
Induction, cycle 1 starting 7 days after reduction |
Prednisone |
PO |
|
Vincristine, high-dose methotrexate with leucovorin rescue, cyclophosphamide, doxorubicin |
IV |
||
Methotrexate/Ara-C/hydrocortisone |
IT |
||
Filgrastim (G-CSF) |
SC or IV |
||
Induction, cycle 2 |
Prednisone |
PO |
|
Vincristine, high-dose methotrexate with leucovorin rescue, cyclophosphamide, doxorubicin |
IV |
||
Methotrexate/Ara-C/hydrocortisone |
IT |
||
Filgrastim (G-CSF) |
SC or IV |
||
Consolidation, 2 cycles† |
High-dose Ara-C, etoposide (VP-16) |
IV |
|
Filgrastim (G-CSF), days 7-21 |
SC or IV |
||
High-dose methotrexate with leucovorin rescue |
IV |
||
Methotrexate/Ara-C/hydrocortisone |
IT |
||
Maintenance 1 |
Prednisone |
PO |
|
Vincristine, high-dose methotrexate with leucovorin rescue, cyclophosphamide, doxorubicin |
IV |
||
Methotrexate/Ara-C/hydrocortisone |
IT |
||
Maintenance 2 |
Ara-C, etoposide (VP-16) |
IT |
|
Maintenance 3 |
Prednisone |
PO |
|
Vincristine, cyclophosphamide, doxorubicin |
IV |
||
Maintenance 4 |
Ara-C, etoposide (VP-16) |
IV |
|
Ara-C = cytarabine; G-CSF = granulocyte colony-stimulating factor; IT = intrathecal; IV = intravenous; PO = oral, SC = subcutaneous * See Table 3 for the definition of group C. † For patients with CNS involvement, during consolidation cycle 1 only. |
|||
Drug |
Route |
Dexamethasone |
PO |
Cyclophosphamide |
IV |
Methotrexate/Ara-C/prednisolone |
IT |
Ara-C = cytarabine; IT = intrathecal; IV = intravenous; PO = oral. |
|
Cycle |
Drug |
Route |
|
A |
Dexamethasone |
PO |
|
Methotrexate with leucovorin rescue, ifosfamide, etoposide (VP-16), Ara-C |
IV |
||
B |
Dexamethasone |
PO |
|
Methotrexate with leucovorin rescue, cyclophosphamide, doxorubicin |
IV |
||
Ara-C = cytarabine; IT = intrathecal; IV = intravenous; PO = oral. |
|||

What would you like to print?
- Overview
- Presentation
- DDx
- Workup
- Treatment
- Approach Considerations
- Tumor Lysis Syndrome Treatment
- Chemotherapy for Lymphoblastic Lymphoma
- Chemotherapy for Small Noncleaved Cell Lymphoma
- Chemotherapy for Large Cell Lymphoma
- Radiation Therapy
- Treatment of Relapsed Disease
- Lymph Node Excision and Dissection
- Consultations
- Long-Term Monitoring
- Show All
- Medication
- Questions & Answers
- Media Gallery
- Tables
- References








