Practice Essentials
Acute myeloid (myelogenous, myelocytic, myeloblastic) leukemia (AML) consists of a group of malignant disorders characterized by the replacement of normal bone marrow with abnormal, primitive hematopoietic cells. Although the cure rate has improved, treatments are associated with notable morbidity and mortality. [1]
Signs and symptoms
Signs and symptoms of pediatric acute myelocytic leukemia (AML) can be divided into the following: (1) those caused by a deficiency of normally functioning cells, (2) those due to the proliferation and infiltration of the abnormal leukemic cell population, and (3) constitutional symptoms.
Symptoms due to a deficiency of normally functioning cells include the following:
-
Cytopenias: Can result from a deficiency of normally functioning cells
-
Anemia: Characterized by pallor, fatigue, tachycardia, and headache
-
Hemorrhage: Most commonly, easy bruising, petechiae, epistaxis, gingival bleeding
-
Fever: Should initially always be attributed to infection
Symptoms due to the proliferation and infiltration of the abnormal leukemic cell mass and infiltrative disease include the following:
-
Extramedullary infiltration: Most commonly in the reticuloendothelial system
-
Mediastinal mass: May cause symptoms of respiratory insufficiency or superior vena cava syndrome
-
Abdominal masses: May cause pain or obstruct the GI or urogenital tracts
-
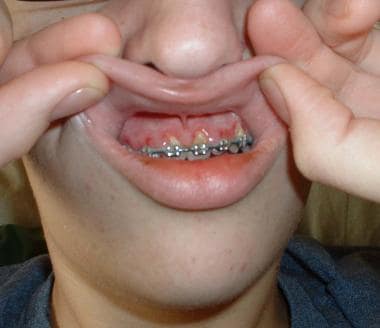
Gingival hyperplasia, CNS infiltration: Often associated with monoblastic leukemia (see the image below)
See Clinical Presentation for more detail.
Diagnosis
Classification of AML
AML can be divided into subtypes on the basis of marrow findings. The French-American-British classification system recognizes 7 primary types of AML (M1-M7), which can usually be established by morphology and additional marrow studies.
The World Health Organization (WHO) classifies AML into groups as follows (rarely used in pediatrics):
-
AML with characteristic cytogenetic translocations
-
AML with multilineage dysplasia
-
AML and myelodysplasia syndromes secondary to therapy
-
AML not otherwise categorized
Testing
The hallmark of AML is the reduction or absence of normal hematopoietic elements. Anemia is usually normocytic, with a lower-than-expected reticulocyte count for the hemoglobin level. The decrease in hemoglobin levels can range from minimal to profound.
Laboratory tests used in patients with AML include the following:
-
Blood counts with differential: WBC counts may be decreased or elevated; platelet counts usually low
-
Blood smears: Primitive granulocyte/monocyte precursors observed; Auer rods present in specimens of circulating blood from many AML patients but particularly prominent in pediatric acute promyelocytic leukemia (APL)
-
Blood chemistries: Frequently elevated serum uric acid, serum muramidase (lysozyme), LDH levels
-
Blood and urine cultures: Always obtain in a child with fever and leukemia
-
Coagulation tests: Perform with initial diagnosis for evidence of DIC indicating APL
-
Histochemical staining: Standard Wright-Giemsa stains and histochemical stains to differentiate the various acute leukemias
Immunophenotyping: To further characterize leukemic cells for different cell lineages and stages of development
-
Cytogenetic testing: To confirm the diagnosis and for prognostic purposes
-
HLA typing: To identify HLA–matched family donors for possible BMT or HSCT in high-risk patients
Imaging studies
Imaging studies are not required for the diagnosis of AML in children, but the following radiologic studies can be helpful in managing complications that arise:
-
Radiography: Routine CXR to rule out mediastinal masses; abdominal images in patients with abdominal pain and distention to rule out perforation; extremity images in patients to rule out metaphyseal bands at the distal femurs (mostly in pediatric ALL), periosteal new bone formation, focal lytic lesions, or pathologic fractures
-
MRI or CT scanning of the head, spine, or other affected areas: For patients with neurologic symptoms to rule out intracranial hemorrhage or infiltrative disease
-
CT scanning of abdomen or sinuses: For abdominal pain or suspected infection of the large bowel; for early detection of asymptomatic sinusitis as cause of persistent, unexplained fevers
-
Echocardiography: To exclude serious infections that affect heart function; also, perform before chemotherapy and periodically with administration of high cumulative doses of anthracyclines (eg, daunomycin, idarubicin)
-
Radionuclide imaging: To detect occult infection that cultures and other imaging modalities do not reveal (eg, occult osteomyelitis, occult deep-tissue infection)
Procedures
-
Bone marrow examination: To establish the diagnosis of AML
-
Lumbar puncture and CSF examination: For diagnostic and therapeutic purposes
See Workup for more detail.
Management
The treatment of AML is directed toward 2 goals: (1) destroying the leukemic cells as rapidly as possible and preventing the emergence of a resistant clone, and (2) supporting the patient through long periods of pancytopenia until their bone marrow achieves hematologic remission and is again producing normal hematopoietic cells.
Pharmacotherapy
Pharmacotherapy used in managing AML includes the following medications:
-
Chemotherapeutic drugs: Cytarabine (cytosine arabinoside), fludarabine, daunorubicin (daunomycin), etoposide, amsacrine, 6-thioguanine, cyclophosphamide, mitoxantrone, tretinoin, arsenic trioxide, L-asparaginase, gemtuzumab ozogamicin, sorafenib, clofarabine
-
Antiemetic drugs: Ondansetron, granisetron, palonosetron, lorazepam, aprepitant, dexamethasone
-
Prophylactic broad-spectrum antimicrobials: Trimethoprim-sulfamethoxazole, penicillin
-
Prophylactic antifungals: Fluconazole, nystatin, voriconazole, caspofungin, micafungin
-
Tumor lysis: Allopurinol, rasburicase
Nonpharmacologic therapy
AML may also be managed with nonpharmacologic treatments such as the following:
-
Allogeneic or autologous BMT following chemotherapy and irradiation: May reduce relapse rates but doesn’t always improve overall survival
-
Radiation treatment: Primarily to treat chloromas and other masses pressing on a vital structure and that may imminently cause irreversible damage; craniospinal irradiation for persistent CNS leukemia
-
Transfusion support: To correct anemia and thrombocytopenia until remission is achieved (eg, RBC transfusions); to correct coagulopathies (FFP)
Surgical options
The role of surgery in AML is limited and may include the following:
-
Placement of a central venous catheter: To begin treatment and to manage all aspects of chemotherapy and transfusion support
-
Biopsy or aspiration of tissue for culture: To detect possible abscess in febrile patients
-
Intervention for an acute abdomen (eg, typhlitis)
See Treatment and Medication for more detail.
Background
Acute myeloid leukemia consists of a group of malignant disorders characterized by the replacement of normal bone marrow with abnormal, primitive hematopoietic cells. If untreated, the disorder uniformly results in death, usually from infection or bleeding. Although the cure rate has improved, treatments are associated with notable morbidity and mortality.
The long-term survival rate for pediatric patients with acute myeloid leukemia is nearly 60%. Acute myeloid leukemia accounts for about 35% of childhood deaths from leukemia. Mortality is a consequence of resistant progressive disease or treatment-related toxicity.
See Acute Myelogenous Leukemia and Pediatric Acute Lymphoblastic Leukemia for complete information on these topics.
Classification of acute myeloid leukemia
Acute myeloid leukemia can be divided into subtypes on the basis of marrow findings. Some of these subtypes have characteristic clinical pictures. The French-American-British classification system recognizes 7 primary types of acute myeloid leukemia (M1-M7), which can usually be established by morphology and additional marrow studies.
The World Health Organization (WHO) has classified acute myeloid leukemias into groups, although this classification is rarely used in pediatrics. However, for general purposes, note the following:
-
Acute myeloid leukemia with characteristic cytogenetic translocations (eg, promyelocytic leukemia with typical t[15;17])
-
Acute myeloid leukemia with multilineage dysplasia
-
Acute myeloid leukemia and myelodysplasia syndromes secondary to therapy (eg, those following alkylating agents)
-
Acute myeloid leukemia not otherwise categorized (eg, erythroid leukemias, monocytic leukemias)
Complications
Immediate and short-term complications include the following:
-
Serious infections
-
Alopecia
-
Emesis
-
GI erosions and bleeding
-
Hemorrhage
-
Malnutrition
-
Nausea
-
Death
Long-term or delayed complications include the following:
-
Congestive heart failure and arrhythmia (rare)
-
Growth and other endocrine disorders
-
Second malignancies
-
Death
Infection
Infection is a major cause of morbidity and mortality in acute myeloid leukemia. Signs of serious infections in children with leukemia are often subtle. Fever at any time must be taken seriously, and appropriate cultures and investigations must be ordered to diagnose and treat it early.
The predisposition to infection is a consequence of granulocytopenia and immunosuppression. The risk of sepsis is greatest when the absolute granulocyte count is less than 200 cells/μL.
Sepsis and pneumonia are particularly common. Causative agents cover the entire gamut of bacterial, fungal, viral, and other pathogens.
Septic shock is most commonly secondary to gram-negative bacteria, Staphylococcus aureus, and group A Streptococcus bacteria and is often lethal.
Because of prolonged neutropenia, immunosuppression, and treatment with broad-spectrum antibiotics, common causes of death are fungal, antibiotic-resistant bacterial, and other opportunistic infections.
Bleeding
Bleeding is the second most common cause of death in acute myeloid leukemia.
Severe GI, pulmonary, or intracranial hemorrhage is frequently observed.
Disseminated intravascular coagulation is a serious potential problem in all patients with acute promyelocytic leukemia (APL) and, to some extent, in those with other acute myelocytic leukemia subtypes. It can occur in association with thrombosis and hemorrhage.
Tumor lysis syndrome
Patients with high leukemic cell counts or massive organomegaly are at significant risk for tumor lysis syndrome.
This condition is often characterized by pronounced metabolic abnormalities, including hyperkalemia, hypocalcemia, hyperuricemia, and renal failure.
Effects of chemotherapy
The aggressive chemotherapy necessary to cure the patient also results in a great deal of morbidity.
Profound myelosuppression due to high-dose, intensive treatment regimens contribute to a high risk of infection and bleeding.
Mucositis and typhlitis in association with intestinal perforation, renal, and pulmonary complications are common problems patients and clinicians face.
Central nervous system complications
Central nervous system (CNS) involvement, with leukemic cell infiltration, hemorrhage, or infection, can cause devastating complications or death.
The risk is particularly high for patients with hyperleukocytosis and white blood cell (WBC) counts of more than 200 X 109/L (>200,000/μL). These patients are at greater risk of intracranial hemorrhage, and their conditions must be treated as true emergencies.
Etiology
Although the cause of acute myeloid leukemia is unknown in most patients, several factors are associated with its development. Despite these correlations, most people exposed to the same factors do not develop leukemia. This pattern suggests that these factors trigger the malignant transformation of cells, perhaps due to the action of one or more oncogenes or tumor suppressor genes. Defects in deoxyribonucleic acid (DNA) repair mechanisms also contribute to the development of acute myeloid leukemia.
Acute leukemia is believed to begin in a single somatic hematopoietic progenitor that transforms to a cell incapable of normal differentiation. Acute myeloid leukemia is a very heterogeneous disease from a molecular standpoint; oncogenic transformation into a leukemic stem cell may occur at different stages of normal hematopoietic cellular maturation, from the most primitive hematopoietic stem cell to later stages, including myeloid/monocytoid progenitor cells and promyelocytes. This determines which subtype of acute myeloid leukemia results, often with very different behavior and growth characteristics.
As opposed to acute lymphoblastic leukemia (ALL), acute myeloid leukemia is most commonly associated with the development of fusion genes resulting from chromosome translocations. Many translocations are characteristic of a particular subtype of acute leukemia and often convey additional prognostic information to the clinician. Although many patients have only a single cytogenetic abnormality, multiple genetic mutations are often required for the complete leukemic transformation.
Many of the leukemic cells no longer possess the normal property of apoptosis, or programmed cell death. As a result, they have a prolonged life span and are capable of unrestricted clonal proliferation. Because transformed cells lack normal regulatory and growth constraints, they have favorable competitive advantage over normal hematopoietic cells. The result is the accumulation of abnormal cells with qualitative defects. The major cause of morbidity and mortality is the deficiency of normally functioning, mature hematopoietic cells rather than the number of malignant cells.
Splenomegaly due to leukemic infiltration may further contribute to pancytopenia by sequestering and destroying circulating erythrocytes and platelets. As the disease progresses, signs and symptoms of anemia, thrombocytopenia, and neutropenia increase.
Leukemic cells may infiltrate other bodily tissues, causing many clinically significant complications, including CNS involvement, pulmonary dysfunction, or skin and gingival infiltration.
Radiation exposure
A great deal of evidence has implicated radiation in leukemogenesis in many patients, as evidenced in Japan after the atomic explosions at Hiroshima and Nagasaki. Although young children had the high risk of developing ALL, teens and adults were most likely to contract acute myeloid leukemia. Most of the leukemias arose within the first 5 years after exposure, although some developed as much as 15 years after exposure.
Reports of increased risk of leukemia among patients who live near nuclear plants are under investigation, but data are lacking. Likewise, early reports that exposure to strong electromagnetic fields is a risk factor for acute leukemia have not been corroborated.
Exposure to toxins and drugs
Exposure to toxic chemicals that cause damage to bone marrow, such as benzene and toluene (used in the leather, shoe, and dry cleaning industries), is associated with leukemia in adults. Direct evidence of this effect in children has not been established. Exposure to pesticides has been noted to increase the risk of acute myeloid leukemia.
A compelling association has been observed after treatment with antineoplastic cytotoxic agents, particularly alkylating agents such as procarbazine, the nitrosoureas, cyclophosphamide, melphalan, and the epipodophyllotoxins etoposide and teniposide. Patients receiving these agents to treat malignancies (eg, Hodgkin disease) have a significantly increased risk of developing a preleukemic syndrome that ultimately transforms into overt acute myeloid leukemia, especially if the agents are administered with radiation therapy.
Genetic factors and syndromes
Children with Down syndrome (trisomy 21) have a 15-fold increased risk of developing leukemia, most commonly acute megakaryoblastic leukemia, compared with the general population. The risk of megakaryoblastic leukemia in Down syndrome is approximately 400 times greater than it is in the rest of the population. Children with Down syndrome who have transient myeloproliferative syndrome as neonates, a condition often indistinguishable from acute leukemia, also have a high risk of developing acute leukemia in subsequent years.
Patients with inherited disorders, such as Shwachman-Diamond syndrome, Bloom syndrome, Diamond-Blackfan anemia, Fanconi anemia, dyskeratosis congenita, and Kostmann syndrome, also have an elevated risk of developing leukemia. Although statistics vary, about 10% of patients with Fanconi anemia, 5-10% of patients with Shwachman-Diamond syndrome, and 1 in 6 patients with Bloom syndrome develop leukemia. The risk of acute myeloid leukemia in patients with dyskeratosis congenita is nearly 200 times that of the normal population. These syndromes share features of poor DNA repair that are believed to predispose affected individuals to leukemogenic stimuli.
Children with neurofibromatosis type I also appear to be at increased risk for developing acute myeloid leukemia.
Although most cases are diagnosed after a relatively brief duration of symptoms, some patients may present with myelodysplasia. This relatively indolent disorder is characterized by slowly progressive anemia or thrombocytopenia. This disorder can be present for many months or even years before it ultimately converts to acute myeloid leukemia.
Epidemiology
United States statistics
Acute myeloid leukemia accounts for nearly 20% of about 3250 newly diagnosed cases of leukemia in children each year. Although 1 in every 3 newly diagnosed leukemias is acute myeloid leukemia, the ratio of acute myeloid leukemia to ALL rapidly decreases until adolescence. [2] During adolescence, the rate increases to account for nearly 50% of all new diagnoses of leukemia.
International statistics
Although leukemia has been reported in children worldwide, the incidence varies widely. In the United States and other highly industrialized countries, acute myeloid leukemia accounts for about 15% of childhood leukemia. In other areas, such as Turkey, nearly one half of children diagnosed with leukemia have acute myeloid leukemia. Childhood leukemia (other than Burkitt type) is less common in Africa, but the ratio of acute myeloid leukemia to ALL is roughly 1:1. Likewise, the incidence of acute myeloid leukemia in Asia is significantly higher than it is in more developed parts of the world, being nearly equal to that of ALL, as reported by Bhatia and Neglia. [3]
Geographic predilection
Minor geographic variations are observed in the incidences of the different subtypes of acute myeloid leukemia. Areas of the world where rates of acute myeloid leukemia are higher than average include Shanghai, New Zealand, and parts of Japan.
Race-, sex-, and age-related demographics
Although ALL is more common in White children than in Black children, acute myeloid leukemia affects all races nearly equally. The incidence of one subtype, APL, is slightly increased in the Hispanic pediatric population. [4]
Male and female distributions are nearly equal at all ages.
Acute myeloid leukemia is diagnosed in persons of all ages, ranging from the newborns to the elderly. In the first year of life, acute myeloid leukemia accounts for nearly one third of all newly diagnosed leukemias. For the rest of the first decade of life, ALL is more common than acute myeloid leukemia by a ratio of 4:1. The incidence of these diseases is roughly equal during adolescence, and the incidence of acute myeloid leukemia increases in adulthood.
Prognosis
With an overall survival rate of 45-60%, the prognosis for children with acute myeloid leukemia has improved significantly since the late 20th century. A Dutch study found that the 5-year survival rate rose from 40% in the early 1990s to 74% in 2010-2015. [5]
A Japanese consortium reported an overall 5-year survival rate of 62%. [6] The long-term, disease-free survival rate is approximately 65% for patients receiving human leukocyte antigen (HLA)–matched stem cell transplants from family donors, but, as with chemotherapy, this rate is lower in high-risk patients. When patients die during treatment or after relapse, the cause is most commonly infection, bleeding, or refractory disease.
A 2012 study from Japan confirmed the results of the AML99 trial for newly diagnosed pediatric patients with AML with a 5-year overall survival (OS) of 75.6% and event-free survival (EFS) of 61.6%. This group compared their results to another cohort of newly diagnosed AML patients and found their results to be the same as the original AML99 trial with 5-year OS of 77.7% and EFS of 66.7%. Interestingly, the 5-year EFS in patients with a normal karyotype was lower compared to the original AML99 trial. [7]
For children with Down syndrome, current outcomes favor younger children, with a survival rate of 84-86% for children younger than age 2 years, 79% for children aged 2-4 years, and only 33% for children older than age 4 years. [8]
Acute promyelocytic leukemia prognosis has an event-free survival rate of 70-80%, with overall survival close to 90%. [9]
A study by Klco et al looked to determine whether genomic approaches can provide novel prognostic information for adult patients with de novo AML. The study found that although comprehensive genomic data from the patients did not improve outcome assessment, the detection of persistent leukemia-associated mutations in at least 5% of bone marrow cells in day 30 remission samples was associated with a significantly increased risk of relapse, and reduced overall survival. [10]
Cytogenetic abnormalities
Leukemia cells demonstrate clonal cytogenetic abnormalities in more than 85% of patients. These changes are often unique to the subtype. For example, the t(15;17) translocation is nearly always found in patients with APL, whereas t(8;21) is most commonly found in those with myeloblastic leukemia.
Some of the cytogenetic abnormalities have now been shown to confer either greater risk of recurrent disease (eg, monosomy 7 and monosomy 5) or lower risk (eg, t[8;21] and inv[16]/t[16;16]).
Molecular studies
In addition to the established prognostic cytogenetic abnormalities, increasing evidence has revealed various molecular abnormalities that have an impact on outcome. The presence of the FLT3/ITD mutation, a receptor tyrosine kinase mutation, has been established as a predictor of worse outcome. These findings on the blast cells are now used to further stratify patients into risk groups with different treatment strategies.
Another gene affecting prognosis is the nucleophosmin (NPM1) mutation. The presence of this mutation has been shown to confer a favorable prognosis for event-free survival, although the combination of NPM1 and FLT3 mutations found in many patients is not favorable.
The presence of MLL gene is usually an unfavorable prognostic marker. The presence of the Wilms tumor gene (WT1) is also an adverse prognostic marker, with patients often failing to achieve complete remission.
Patient Education
Family members should be familiar with signs of infection other than fever. Dermatologic clues of bleeding, especially petechiae and purpura, should be recognized and investigated.
Discuss the adverse effects of chemotherapy and transplantation at length with family members.
Psychosocial intervention is often necessary for the patient and his or her parents and siblings. A diagnosis of leukemia has profound effects on all family members, with a dramatic change in the patient's lifestyle until all treatment is completed.
Home tutoring is often necessary during the entire period of treatment.
For patient education information, see Leukemia.
-
Gingival hyperplasia in a patient with monoblastic leukemia.
-
Leukemia cutis (a skin nodule) in a patient with leukemia.
Tables

What would you like to print?
- Overview
- Presentation
- DDx
- Workup
- Blood Counts and Blood Smears
- Blood Chemistries and Other Blood Work
- Radiography
- CT Scanning and MRI
- Ultrasonography
- Radionuclide Imaging
- Histochemical Staining
- Immunophenotyping
- Cytogenetics
- HLA Typing
- Bone Marrow Examination
- Histologic Findings
- Lumbar Puncture and Cerebrospinal Fluid Examination
- Show All
- Treatment
- Approach Considerations
- Chemotherapy
- Postinduction Therapy
- Treatment of Acute Promyelocytic Leukemia
- Treatment in Children With Down Syndrome
- Radiation Therapy
- Blood and Marrow Transplantation
- Transfusion Support
- Metabolic Management
- Antibiotic Therapy
- Treatment With Biologic-Response Modifiers
- Surgical Care
- Dietary Modification
- Activity Restrictions
- Deterrence/Prevention of Acute Myelocytic Leukemia
- Monitoring and Follow-Up Care
- Consultations
- Show All
- Medication
- Questions & Answers
- Media Gallery
- References




