Background
Long-chain 3-hydroxyacyl-CoA dehydrogenase (LCHAD) is 1 of 3 enzymatic activities that make up the trifunctional protein of the inner mitochondrial membrane. The other 2 activities of the protein are 2-enoyl coenzyme A (CoA) hydratase (LCEH) and long-chain 3-ketoacyl CoA thiolase (LCKT). The protein is an octamer composed of 4 alpha subunits that contain the LCEH and LCHAD activities and 4 beta subunits that contain the LCKT activity. This enzyme complex metabolizes long-chain fatty acids, and LCHAD activity is specific for compounds of C12-C16 chain length. The genes for the alpha and beta subunits have been localized to chromosome 2. The HADHA gene has been cloned, and a common mutation, c.1528G>C, has been identified in the mutant alleles of LCHAD deficiency. [1]
LCHAD deficiency is a severe fatty acid oxidation disorder that is fatal if untreated. [2] Infants with LCHAD deficiency, which is inherited as an autosomal recessive trait, present in infancy with acute hypoketotic hypoglycemia. [3, 4] These episodes typically appear for the first time after a fast, which usually occurs in the context of intercurrent illness with vomiting.
Pathophysiology
The molecular defect occurs in the mitochondrial trifunctional protein (MTP). Some patients who are deficient in all 3 enzymatic activities of the protein have been described, although most have an isolated LCHAD deficiency, which results in the inability to metabolize long-chain fatty acids. Thus, the clinical features may result from either toxicity due to long-chain acyl-CoA esters that cause cardiomyopathy and cardiac arrhythmias or from a block in long-chain fatty acid oxidation that leads to an inability to synthesize ketone bodies and/or adenosine triphosphate from long-chain fatty acids. See the image below.
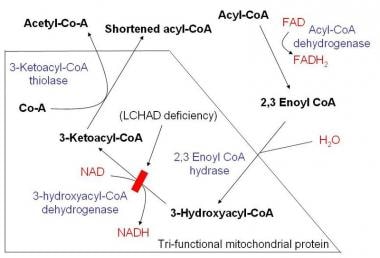 Schematic demonstrating mitochondrial fatty acid beta-oxidation and effects of long-chain acyl CoA dehydrogenase deficiency (LCHAD) deficiency.
Schematic demonstrating mitochondrial fatty acid beta-oxidation and effects of long-chain acyl CoA dehydrogenase deficiency (LCHAD) deficiency.
It has been suggested that mitochondrial energy and Ca2+ homeostasis disruptions caused by the predominant accumulation of the long-chain hydroxyl fatty acid (LCHFA) may contribute to the severe cardiac and hepatic clinical features, [5] muscular symptoms, and recurrent rhabdomyolysis [6] in patients with LCHAD deficiency. Another study confirmed that disturbance of mitochondrial functions caused by oxidative stress from the accumulating fatty acids is involved in the pathophysiology of LCHAD deficiency. This pathomechanism may play a role in chronic and neurologic symptoms of LCHAD deficiency. [7]
Increased rates of lipolysis after fasting have been observed. [8] The increased lipolysis may represent a compensatory mechanism to meet energy demands after few hours of fasting. However, this effect may be achieved at the cost of fatty acid infiltration and of toxic effects of β-oxidation intermediates on organ functions. Patients with LCHAD deficiency may develop a profound CNS deficiency of docosahexanoic acid ethyl ester (DHA), 22:6n-3. An association between retinopathy and DHA deficiency has been demonstrated. The etiology of the severe peripheral neuropathy of trifunctional protein deficiency may result from the unique metabolite, 3-keto-acyl-CoA, after conversion to a methylketone via spontaneous decarboxylation.
The fatty acid oxidation defect results in adverse effects on numerous organ systems, including the CNS, secondary to the hypoketotic hypoglycemia. Hypotonia and cardiomyopathy are also usually present, reflecting the underlying energy deficiency. In addition, hepatomegaly is usually evident, and biopsy of the liver reveals fat accumulation and fibrosis. Chorioretinopathy may also develop over time. Chronic hemolytic anemia and delayed CNS myelination have also been reported. [9]
Epidemiology
Frequency
United States
The incidence of isolated LCHAD activity deficiency and trifunctional protein deficiency is unknown in the United States.
International
Analysis of the frequency of the most common mutation (c.1528G>C) in the HADHA gene that encodes for mitochondrial LCHAD estimated a carrier frequency of 1:240 in Finland. [10] This mutation was also present in 100% of alleles in patients in Ukraine, and, in 2018, the frequency of LCHAD deficiency in Ukraine was found to be 1:329,968, which is 2.1 times lower than the average in Europe. [11] A significantly higher frequency of the same mutation was found in the adult Kashubian population from North Poland. [12] The same study also found a higher frequency of another polymorphism, c.652G>C, in the HADHA gene within the population of Silesian in southern Poland. Fourteen living patients with LHAD deficiency were reported in Austria in 2015 and were all homozygous for the common mutation c.1528G>C. [13]
Mortality/Morbidity
In most cases, LCHAD deficiency is severe and may lead to death during the first few months of life. The disease may also be a cause of sudden infant death, even neonatal. Infants who are diagnosed and treated still have a risk for psychomotor retardation. In addition, this homozygous mutation in fetuses was found to be highly associated with maternal acute fatty liver of pregnancy. [14]
Race
LCHAD deficiency has been reported in all ethnic groups.
Sex
LCHAD deficiency has no sexual predilection because it is an autosomal recessive disorder.
Age
Patients with LCHAD activity deficiency usually present with hypoketotic hypoglycemia, cardiomyopathy, hypotonia, and hepatomegaly at a median age of 6 months. In childhood, the presentation is myopathic. A minority of patients (up to 15%) may present during the neonatal period. A late-onset neuromuscular disease has been reported in MTP deficiency.
Prognosis
In LCHAD deficiency, the fulminant acute symptoms may be difficult to manage and resistant to therapeutic attempts (with high mortality) because the presentations may involve a lethal acute liver failure, a rapidly evolving cardiomyopathy, or hypoketotic hypoglycemic encephalopathy. However, treatment may improve the long-term prognosis.
Conventional therapy may not be sufficient to prevent ophthalmological changes; however, early diagnosis and adequate therapy may delay the progression of retinal complications.
Patient Education
Advise family members to learn cardiopulmonary resuscitation (CPR).
Teach family members to recognize signs and symptoms of hypoglycemia and instruct them to provide oral sources of glucose, glucose gel, or glucagon injection while waiting for emergency aid.
Educate family members about frequent feeds and avoidance of fasting in general. If decreased oral intake occurs, the child should be seen immediately at the pediatrician's office or rushed to the emergency department.
Educate the family about the importance of a fat-restricted high-carbohydrate diet with MCT oil supplementation and use of uncooked cornstarch to prevent episodes of hypoglycemia (see Diet).
Provide education about routine ophthalmological follow-up care to screen for the onset of pigmentary retinopathy.
Educate the family about pregnancy complications mainly described in heterozygous mothers giving birth to affected fetuses (eg, HELLP syndrome, acute fatty liver of pregnancy).
Arrange for genetic counseling and discussion of recurrence risk for future pregnancies.
Educate about the possibility of prenatal diagnosis, which may be performed by measuring acylcarnitine profiles, measuring the activity of specific enzymes, or by searching for identified mutations (G1528C) from amniocytes or chorionic villus cells.
Provide education about carnitine supplementation if significant hypocarnitinemia during the asymptomatic state is documented.
-
Schematic demonstrating mitochondrial fatty acid beta-oxidation and effects of long-chain acyl CoA dehydrogenase deficiency (LCHAD) deficiency.



