Background
Insulin-like growth factor I (IGF-I) is the effector of growth induced by growth hormone (GH). IGF-I deficiency can be the result of GH resistance or insensitivity due to genetic disorders of the GH receptor causing GH receptor deficiency (growth hormone receptor deficiency [GHRD], Laron syndrome) or postreceptor defects, including the principal transduction agent STAT5b, the IGF-I/IGFBP3 stabilizer acid labile subunit (ALS), the IGF-I gene, or the IGF-I receptor. [1]
Acquired forms of GH insensitivity include the rare GH1 mutation (in which GH inhibiting antibodies develop after a few months of replacement therapy with recombinant GH) and, far more commonly, malnutrition, hepatic disease, renal disease, and diabetes. The table below compares the clinical and biochemical features associated with these various causes of GH resistance.
Table. Features of GH Resistance Causes (Open Table in a new window)
Condition |
Growth failure |
GH |
GH binding protein |
IGF-I |
IGFBP3 |
Genetic |
|||||
GHRD - Recessive forms |
Severe |
Elevated |
Absent-low* |
Very low |
Very low |
GHRD - Dominant negative forms |
Mild-moderate |
Elevated |
Increased |
Very low |
Low-normal |
STAT5b mutation |
Severe |
Elevated |
Normal |
Very low |
Very low |
ALS mutation |
None-moderate |
Normal |
Normal |
Very low |
Very low |
IGF-I gene mutation |
Severe |
Elevated |
Normal |
Absent-high** |
Low-normal |
IGF-I receptor mutation |
Mild-moderate |
Normal-elevated |
Normal |
Normal-elevated |
Normal-elevated |
Acquired |
|||||
GH inhibiting antibodies |
Severe |
Absent |
Normal |
Very low |
Low |
Malnutrition |
None-mild |
Elevated |
Decreased |
Variable |
Variable |
Diabetes mellitus |
None-mild |
Elevated |
Decreased |
Decreased |
Increased |
Renal disease |
Mild-severe |
Normal |
Decreased |
Normal |
Increased |
Hepatic disease |
Mild-severe |
Elevated |
Normal-increased |
Decreased |
Normal |
*Increased in mutations of or near the transmembrane domain of the GH receptor**Absent with partial IGF1 gene deletion; very high with abnormal IGF-I |
|||||
Pathophysiology
The GH molecule binds to its specific cell surface receptor (GHR), which dimerizes with another GHR molecule so that the single GH molecule is enveloped by 2 GHR molecules. The intact receptor lacks tyrosine kinase activity, but binding of GH and dimerization results in association with JAK2, a member of the Janus kinase family, which results in self-phosphorylation of the JAK2 and a cascade of phosphorylation of cellular proteins. The most critical of these proteins is the signal transducer and activator of transcription 5b (STAT5b), which couples GH binding to the activation of gene expression that leads to the intracellular effects of GH, including synthesis of IGF-I, insulin-like growth factor binding protein 3 (IGFBP3), and ALS. [1]
Hepatic IGF-I circulates almost entirely bound to IGF binding proteins (IGFBPs), with less than 1% being free. The IGFBPs are a family of 6 structurally related proteins with a high affinity for binding IGF. The principal BP, IGFBP3, binds approximately 90% of circulating IGF-I in a large (150-200 kD) ternary complex consisting of IGFBP3, ALS, and the IGF molecule. The ALS stabilizes the IGF–IGFBP3 complex, reduces the passage of IGF-I to the extravascular compartment, and extends its half-life. [1]
IGF binding involves 3 basic types of receptors: the structurally homologous insulin receptor and type 1 IGF receptor and the distinctive type 2 IGF-II/mannose-6-phosphate receptor. In addition to these receptors, hybrid receptors consisting of a dimer from the IGF-I receptor paired with the insulin receptor, are ubiquitous and the respective expression of these receptors varies from tissue to tissue. Although the insulin receptor has a low affinity for IGF-I, IGF-I is present in the circulation at molar concentrations that are 1000 times those of insulin. Thus, even a small insulin-like effect of IGF-I could be more important than that of insulin itself, were it not for the IGFBPs that control the availability and activity of IGF-I. In fact, intravenous infusion of recombinant human IGF-I (rhIGF-I) can induce hypoglycemia, especially in the IGFBP3 deficient state. [2]
The importance of IGF-I in normal intrauterine growth in humans has been demonstrated in a single patient with a homozygous partial deletion of the IGF1 gene, [3] and patients with mutation of the IGF1 gene resulting in high circulating levels of an ineffective IGF-I, [4, 5, 6] and in probands and first-degree relatives with heterozygous mutations of the IGF-I receptor. [7] Those individuals with IGF1 gene mutations were severely mentally retarded and had growth retardation at birth, indicating dependence of both intrauterine somatic growth and brain development on adequate IGF-I. Those with heterozygous mutations of the IGF-I receptor had more moderate intrauterine growth retardation and inconsistent mental retardation. Individuals with mutations of STAT5b do not appear to have intrauterine growth retardation or impaired brain development; however, because of the central role of STAT5b in multiple cytokine transduction/transcription pathways, these individuals can have serious immunodeficiency problems. [7]
Epidemiology
United States data
Very few of the reported cases of GH resistance due to GHRD or the even more rare postreceptor abnormalities come from North America.
International data
Worldwide, approximately 250 individuals have GHRD/Laron syndrome, 10 individuals have homozygous STAT5b mutations, 13 families have IGF-I receptor mutations affecting over 20 individuals, and a comparable number of individuals have homozygous mutations resulting in ALS deficiency; only 3 individuals have been reported with IGF1 gene mutations. Recombinant IGF-I treatment reports include 13 children with GH gene deletion and acquired GH inhibiting antibodies following rhGH therapy. Other forms of acquired GH resistance, due to malnutrition or chronic disease, are common.
Race-related demographics
Among the approximately 250 affected individuals with GHRD identified worldwide, about two thirds are Semitic and half of the rest are of Mediterranean or South Asian origin. The Semitic group includes the Arab, Oriental, or Middle Eastern Jewish population and the largest group, the genetically homogeneous 100+ Conversos in Ecuador (Jews who converted to Christianity during the Inquisition).
The identification of an Israeli GHRD patient of Moroccan origin with the E180splice mutation found in the Ecuadorian patients indicated the Iberian provenance of this mutation, which readily recombined in the isolated communities of these 16th century immigrants established in the southern Ecuadorian Andes. Recently, additional patients with the E180splice mutation on the same genetic background have been identified in Chile and Brazil and in siblings from Mexico residing in the US, indicative of a common founder. [1]
The 10 individuals with STAT5b mutation include Kuwaiti siblings, 2 related and 2 unrelated Argentinians, 1 patient from Turkey, and 2 siblings from Brazil. [11]
ALS mutations were reported in Kurds, several unrelated Spanish patients, Norwegian/German siblings, and patients of Turkish, Argentinian, Ashkenazi Jewish, Pakistani, mixed European, and Mayan origin. [13, 14, 15, 16, 17, 18, 19]
Families with mutations of the IGF-I receptor were of Dagestani, European, and Japanese origin. [7, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30]
Sex-related demographics
Among patients observed from the original description of GHRD by Laron, Pertzelan, and Mannheimer in 1966 [31] until 1990, a normal sex ratio was noted. The initial report of 20 cases from a single province in Ecuador included only 1 male, but subsequent observations from an adjacent province indicated a normal sex ratio, and a few more males were subsequently identified in the initial province. [8, 32] The abnormal sex ratio for that locus (M/F = 1/4) remains unexplained.
Age-related demographics
Newborns with GHRD are instantly recognizable to family members with previous experience because of the foreshortened facies and prominent brow. This is a curious finding, because intrauterine growth is not dependent on GH-GHR interaction.
Prognosis
Long-term prognosis appears normal for GHRD, and postreceptor defects with the exception of STAT5b. In fact, subjects with GHRD, despite obesity, have enhanced insulin sensitivity and do not develop diabetes and may be protected from cancer, as well. [33]
Individuals with STAT5b mutation probably have a reduced life expectancy as a result of chronic pulmonary disease.
Mortality/Morbidity
Children younger than 7 years with GHRD had twice the mortality of their unaffected siblings in a large Ecuadorian cohort, with causes of death not being different and typical of the environment (meningitis, diarrhea, pneumonia). [8]
Lean to fat mass ratios determined by dual energy x-ray absorptiometry are markedly reduced in studies of Ecuadorian and Israeli patients (who together account for more than half of known cases). [8, 9] Total and LDL cholesterol levels are elevated, likely reflecting decreased activity of the hepatic LDL receptor that is under direct GH influence.
Longevity after early childhood appears normal, although as with GH deficiency, there is an old-young appearance due to wrinkling and sagging of the face.
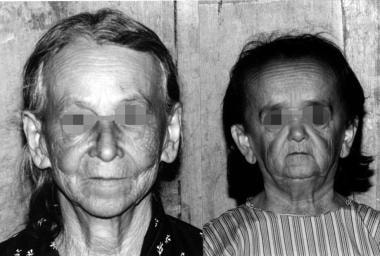 A 50-year-old woman with GHRD (right) and her 75-year-old mother, indicating premature aging appearance. Photos were taken at the same distance, emphasizing the small size of the subject and relative foreshortening of the facies.
A 50-year-old woman with GHRD (right) and her 75-year-old mother, indicating premature aging appearance. Photos were taken at the same distance, emphasizing the small size of the subject and relative foreshortening of the facies.
At least 50% of infants and children with GHRD have overt symptoms of hypoglycemia, including convulsions, and many without a clinical history of symptoms demonstrate quite low blood glucose levels with ordinary fasting. Retardation associated with severe recurrent hypoglycemia has only been noted in one instance. A somewhat increased mental retardation rate of 13.5% in an international treatment study series, and wide variability of intellectual capabilities in the Israeli population with GHRD that did not correlate with hypoglycemia histories, are observations that are not controlled by concurrent studies of unaffected family members, and they likely reflect the frequent association of this disorder with consanguinity. Controlled studies in the Ecuadorian cohort failed to demonstrate intellectual impairment or impaired school performance. [10]
 A 10-year-old Ecuadorian girl with GHRD/Laron syndrome, who was performing at the top of the class, with her classmates.
A 10-year-old Ecuadorian girl with GHRD/Laron syndrome, who was performing at the top of the class, with her classmates.
The ALS and STAT5b mutations are not associated with intellectual impairment, because, as with GHRD (and as has long been known with congenital GH deficiency), intrauterine IGF-I production is not impaired and presumably GH independent. As noted earlier, IGF1 gene mutations result in severe mental retardation, and heterozygous IGF-I receptor mutations have no retardation to moderate retardation.
Because the STAT5b pathway is important in immune function, 9 of 10 reported individuals with functional mutation of the STAT5b have had severe immunologic problems or chronic pulmonary disease; the initially reported patient was awaiting lung transplantation. [11]
Reproductive capability has been normal in the GHRD population. Women require cesarean delivery.
Patient Education
With IGF-I therapy, parents and patients are instructed to observe for and report adverse effects. Parents and patients are also instructed to recognize the signs and symptoms of hypoglycemia, how to treat hypoglycemia, and, when deemed appropriate, how to monitor blood sugar at home.
For patient education resources, see the Thyroid & Metabolism Center and Oral Health Center, as well as Growth Hormone Deficiency, Growth Hormone Deficiency in Children, Growth Failure in Children, Growth Hormone Deficiency FAQs, and Growth Hormone Deficiency Medications.
-
Diagram of the hypothalamic-pituitary-GH/IGF-I axis, showing mutational targets beginning with the GH-releasing hormone receptor (GHRHR), indicated in bold and italicized.
-
A 50-year-old woman with GHRD (right) and her 75-year-old mother, indicating premature aging appearance. Photos were taken at the same distance, emphasizing the small size of the subject and relative foreshortening of the facies.
-
A 10-year-old Ecuadorian girl with GHRD/Laron syndrome, who was performing at the top of the class, with her classmates.
-
15 Ecuadorian children with GHRD due to homozygosity for the E180 splice mutation of the GH receptor, lined up according to descending age from 15 years to 2 years, with 3 normal children standing behind age mates. Note general but not consistent statural correlation with age, most dramatic for the 11-year-old boy, 4th from the left, and his 8-year-old brother holding the ball who is almost the same height.
-
A 21-year-old woman and her 23-year-old brother with GHRD/Laron syndrome demonstrating variable effects on growth of the same mutation and the correlation with low levels of IGF-I in IGFBP3. Her height is 100 cm, -11.2 SDS and his height is 134 cm, -6.3 SDS, his IGF-I level is 4 times hers, and his IGFBP3 level is twice hers.
-
Adult with GHRD standing with 3 of his fellow police officers, his affected brother, a visiting US physician (Dr Frank Diamond) and the seated chief.
-
Six-month, placebo-controlled, double-blind study of rhIGF-I in 16 Ecuadorian children with GHRD, followed by 6 months open label rhIGF-I therapy of the entire group.
-
Treatment with rhIGF-I for 1-2 year of children with GH insensitivity. Data are from the references noted as well as package inserts.
-
Four subjects with growth hormone (GH) receptor deficiency due to the E180 splice mutation on the GH receptor gene. From left to right, the first woman, age 22 years, was treated from age 4 years, when she had a height standard deviation score (SDS) of -8, to age 14 years with insulinlike growth factor-1 at a dose of 80 µg/kg body weight bid; adult height is -4.3 SDS and body fat percent is 39.8. The other 3 women were treated for 3 years with 120 µg/kg bid and are aged 30, 23, and 27 years with body fat content of 49.3%, 49%, and 54.6% and with heights of 120.7 cm, 120.8 cm, and 118.5 cm, respectively. Females with GH insufficiency who had comparable baseline characteristics and were treated with 120 µg/kg twice daily to adult height in the US trial only reached 112 cm, 121.2 cm, and 120.8 cm. These observations suggest no greater statural attainment with prolonged high-dose therapy than with short-term, high-dose treatment, consistent with the observation of disproportionate advancement of osseous maturation by the higher dose. Courtesy of The Journal of Clinical Endocrinology and Metabolism (Guevara-Aguirre J, Rosenbloom AL, Guevara-Aguirre M, Saavedra J, Procel P. Recommended IGF-I dosage causes greater fat accumulation and osseous maturation than lower dosage and may compromise long-term growth effects. J Clin Endocrinol Metab 98: 839–845, 2013).
Tables
Condition |
Growth failure |
GH |
GH binding protein |
IGF-I |
IGFBP3 |
Genetic |
|||||
GHRD - Recessive forms |
Severe |
Elevated |
Absent-low* |
Very low |
Very low |
GHRD - Dominant negative forms |
Mild-moderate |
Elevated |
Increased |
Very low |
Low-normal |
STAT5b mutation |
Severe |
Elevated |
Normal |
Very low |
Very low |
ALS mutation |
None-moderate |
Normal |
Normal |
Very low |
Very low |
IGF-I gene mutation |
Severe |
Elevated |
Normal |
Absent-high** |
Low-normal |
IGF-I receptor mutation |
Mild-moderate |
Normal-elevated |
Normal |
Normal-elevated |
Normal-elevated |
Acquired |
|||||
GH inhibiting antibodies |
Severe |
Absent |
Normal |
Very low |
Low |
Malnutrition |
None-mild |
Elevated |
Decreased |
Variable |
Variable |
Diabetes mellitus |
None-mild |
Elevated |
Decreased |
Decreased |
Increased |
Renal disease |
Mild-severe |
Normal |
Decreased |
Normal |
Increased |
Hepatic disease |
Mild-severe |
Elevated |
Normal-increased |
Decreased |
Normal |
*Increased in mutations of or near the transmembrane domain of the GH receptor**Absent with partial IGF1 gene deletion; very high with abnormal IGF-I |
|||||






