Practice Essentials
In 1942, Klinefelter et al published a report describing nine men with a constellation of features: testicular dysgenesis, microorchidism, eunuchoidism, gynecomastia, elevated urinary gonadotropins, and azoospermia. [1] The etiology was thought to be due to an endocrine disorder of unknown cause, until 1959, when Jacobs et al recognized that Klinefelter syndrome was a chromosomal disorder in which there is an extra X chromosome, resulting in the karyotype 47,XXY. [2]
Today, the term Klinefelter syndrome (KS) refers to a group of chromosomal disorders in which the normal male karyotype, 46,XY, has at least one extra X chromosome. XXY aneuploidy, the most common human sex chromosome disorder, has a prevalence of 1 in 500 males. [3] It is also the most common chromosomal disorder associated with male hypogonadism and infertility.
Other sex chromosomal aneuploidies are included in the KS group of chromosomal disorders. Arising less frequently, 48,XXYY and 48,XXXY occur in 1 per 17,000 to 50,000 male births, while 49,XXXXY has an incidence of 1 per 85,000 to 100,000 male births. [3]
Signs and symptoms of Klinefelter syndrome
Klinefelter syndrome is characterized by hypogonadism (micro-orchidism [small testes], oligospermia/azoospermia), gynecomastia in late puberty, hyalinization and fibrosis of the seminiferous tubules, elevated urinary gonadotropin levels, and behavioral concerns.
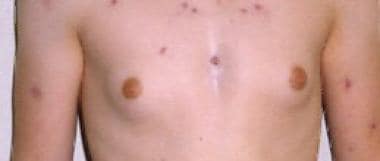
Images of phenotypic features seen in Klinefelter syndrome are shown below:
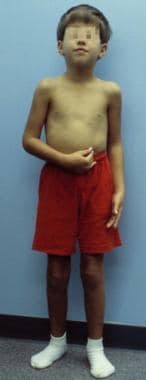 Child with Klinefelter syndrome. Other than a thin build and disproportionately long arms and legs, the phenotype is normal.
Child with Klinefelter syndrome. Other than a thin build and disproportionately long arms and legs, the phenotype is normal.
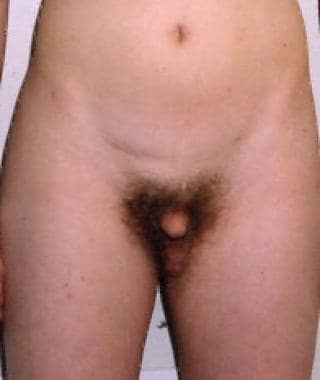 Adolescent male with Klinefelter syndrome who has female-type distribution of pubic hair and testicular dysgenesis.
Adolescent male with Klinefelter syndrome who has female-type distribution of pubic hair and testicular dysgenesis.
Workup in Klinefelter syndrome
Klinefelter syndrome may be diagnosed prenatally from fetal cytogenetic analyses performed on chorionic villi or amniocytes. If Klinefelter syndrome is not diagnosed prenatally, a patient with 47,XXY karyotype may demonstrate various subtle, age-related clinical signs that would prompt diagnostic testing. Karyotype analysis on peripheral blood lymphocytes, the XCAT-KS buccal swab test, fluorescence in-situ hybridization (FISH), and microarrays are options for postnatal diagnostic testing.
Management
Early identification and anticipatory guidance are extremely helpful in Klinefelter syndrome. Management and treatment should focus on 3 major facets of the syndrome: hypogonadism, gynecomastia, and psychosocial problems. Androgen (testosterone) replacement therapy is an important aspect of treatment.
A multidisciplinary team approach can assist in improving speech impairments, academic difficulties, and other psychosocial and behavioral problems.
Physical therapy is recommended for boys with hypotonia or delayed gross motor skills that may affect muscle tone, balance, and coordination. Occupational therapy is advised in boys with motor dyspraxia.
Until 1996, men with Klinefelter syndrome were considered infertile. Since then, however, developments in microsurgical techniques and advances in artificial reproductive technologies (ART) have enabled over 50% of men with Klinefelter syndrome to sire their own children. [4, 5, 6, 7, 8]
Pathophysiology
The X chromosome carries genes that have a role in many organ systems, playing a part, for example, in testes function, brain development, and growth. [9] Consequences of an extra X chromosome, usually acquired through a nondisjunctional error during parental gametogenesis, include hypogonadism, gynecomastia, and psychosocial behavioral concerns.
The addition of more than one extra X or Y chromosome to a normal male karyotype results in variable cognitive and physical abnormalities. As the number of supernumerary X chromosomes increases, somatic and cognitive development are more likely to be affected. Skeletal and cardiovascular abnormalities can become increasingly severe. Gonadal development is particularly susceptible to each additional X chromosome, resulting in seminiferous tubule dysgenesis and infertility, as well as hypoplastic and malformed genitalia, as seen in polysomy X males. A form of primary testicular failure occurs in males with Klinefelter Syndrome, with elevated gonadotropin levels due to lack of feedback inhibition by the pituitary gland.
Moreover, mental capacity diminishes with additional X chromosomes. The intelligence quotient (IQ) score is reduced by approximately 15 points for each supernumerary X chromosome, but conclusions about reduced mental capacity must be drawn cautiously. All major areas of development, including expressive and receptive language and coordination, are affected by extra X chromosome material.
A study by van Rijn et al indicated that information processing becomes more difficult in individuals with a 47,XXY karyotype as the social load related to the task increases. For example, with regard to visuospatial processing, which was associated with no social load, 17% of subjects in the study had trouble with the task, while in terms of facial recognition (medium social load) and facial expression of emotion (high social load), 26% and 33% of individuals, respectively, encountered difficulties. [10]
Androgen deficiency causes the following:
-
Eunuchoid body proportions/female distribution of adipose tissue
-
Sparse or absent facial, axillary, pubic, or body hair
-
Gynecomastia
-
Decreased muscle mass and strength
-
Decreased physical endurance
-
Small testes and penis
-
Diminished libido
-
Loss of functional seminiferous tubules and Sertoli cells, causing decrease in inhibin B levels (the hormone regulator of the follicle-stimulating hormone [FSH] level)
-
Altered hypothalamic-pituitary-gonadal axis in pubertal boys
Men with Klinefelter syndrome have a higher risk of autoimmune diseases, diabetes mellitus and its associated complications, osteopenia and osteoporosis, tumors (breast and germ cells), systemic lupus erythematosus, rheumatoid arthritis, and Sjögren syndrome. [11, 12] This higher risk is comparable to the disease risk for 46,XX females.
Sera laboratory results of a typical male with Klinefelter syndrome demonstrate low to low-normal testosterone levels, high luteinizing hormone (LH) and FSH levels, and, often, elevated estradiol levels. Decline of testosterone production progresses over the patient's life span, but not all male patients have hypogonadism. [13] It is unclear if the morbidity associated with Klinefelter syndrome is a result of hypogonadism and hyperestrogenism or is due to abnormal function of X chromosome–linked genes. [6]
A study by Close et al reported that in boys with Klinefelter syndrome, the degree of phenotypic abnormality is tied to the risk for impaired quality of life. Linear regression analysis indicated that phenotype accounted for 22% of the variance in quality of life among the 43 boys in the study. [14]
Using contrast-enhanced ultrasonography, a study by Carlomagno et al indicated that testicular venous flow in patients with Klinefelter syndrome is a predictor of total testosterone levels. Comparing eugonadal males with Klinefelter syndrome patients, the investigators found that the latter showed prolongation of wash-in time, mean transit time, time to peak, and wash-out time. Moreover, an association was seen between faster testicular blood flow and higher total testosterone levels. [15]
A Danish survey report, by Skakkebæk et al, indicated that persons with Klinefelter syndrome tend to have reduced mental and physical quality of life, caused directly by Klinefelter syndrome and indirectly by factors that, compared with controls, include lower levels of income, physical activity, and sexual function. [16]
Epidemiology
Frequency
United States
Klinefelter syndrome (XXY aneuploidy) is the most common human sex chromosome disorder.
-
Approximately 1 in 500-600 males is born with an extra X chromosome.
-
The prevalence rate is 5-20 times higher in males who are mentally challenged than in the general male population.
-
Approximately 250,000 men in the United States have Klinefelter syndrome. [17]
Mortality/Morbidity
About 40% of concepti with Klinefelter syndrome survive in utero to the postnatal period.
-
In general, the severity of somatic malformations in Klinefelter syndrome is proportional to the number of supernumerary X chromosomes; intellectual disability and hypogonadism are more severe in patients with 49,XXXXY than in those with 48,XXXY.
-
The mortality rate is not significantly higher than in healthy individuals.
Race
Klinefelter syndrome does not have any racial predilection.
Sex
Klinefelter syndrome (47,XXY) results from an additional X chromosome on an XY background; therefore, this condition affects only males.
Age
Klinefelter syndrome is often undiagnosed in young males. Diagnosis frequently occurs in adulthood; however about 75% of sex chromosome aneuploidies are never diagnosed. For suspected 47,XXY males, common indicators for karyotype analysis on peripheral blood are hypogonadism and infertility.
-
Adolescent male with gynecomastia and Klinefelter syndrome.
-
Child with Klinefelter syndrome. Other than a thin build and disproportionately long arms and legs, the phenotype is normal.
-
G-banded 47,XXY karyotype.
-
Adolescent male with Klinefelter syndrome who has female-type distribution of pubic hair and testicular dysgenesis.






