Background
Kernicterus, or bilirubin encephalopathy, is bilirubin-induced neurologic damage, typically in infants. [1] The term kernicterus literally means "yellow kern," with kern indicating the most commonly afflicted region of the brain (ie, the nuclear region). Historically, the term refers to an anatomic diagnosis made at autopsy based on a characteristic pattern of staining found in babies who had marked hyperbilirubinemia before they died.
Hervieux first described the condition in 1847, and Schmorl first used the term kernicterus as early as 1903. Regions most commonly affected include the basal ganglia; hippocampus; geniculate bodies; and cranial nerve nuclei, such as the oculomotor, vestibular, and cochlear. The cerebellum can also be affected. Bilirubin-induced neurologic dysfunction (BIND) refers to the clinical signs associated with bilirubin toxicity (ie, hypotonia followed by hypertonia and/or opisthotonus or retrocollis) and is typically divided into acute and chronic phases. The two terms are commonly used interchangeably, but this use is not technically accurate because one refers to clinical manifestations and the other to an anatomic diagnosis.
Conventional wisdom characterizes kernicterus as prevalent in the 1950s and 1960s, virtually eradicated in the 1970s and 1980s, only to reappear during the 1990s. It was speculated that early discharge of term infants (before their serum bilirubin concentration peaks) could be a factor in the reemergence of this devastating neurologic affliction, and medical research focused on developing surveillance and treatment paradigms to eliminate the condition. Whereas it is undeniable that kernicterus remains a cause of major neurologic morbidity in the infant population, population studies of children born in California between 1988 and 1997 suggest the prevalence of kernicterus has remained virtually unchanged since 1980. [2]
Much of the traditional teaching regarding hyperbilirubinemia has been questioned as more is learned about bilirubin metabolism and neurologic injury. Kernicterus is now recognized in the premature infant and, very rarely, in the term infant in the absence of profound hyperbilirubinemia [3] ; however, other problems (eg, acidosis or infection) are present in term infants without profound hyperbilirubinemia. Conversely, physiologic jaundice (sometimes to levels previously thought to be universally dangerous) has been recognized to be within the reference range in the first week of life in healthy term babies, particularly those who are breastfed. Jaundice of this type usually spontaneously resolves without sequelae.
The lack of a clear correlation between the measured bilirubin level and the development of kernicterus continues to confuse clinicians and confound preventive measures. [4] Despite the lack of a clear-cut cause-and-effect relationship between kernicterus and the degree of hyperbilirubinemia, laboratory investigations have demonstrated that bilirubin is neurotoxic at a cellular level. Other in vitro studies have shown bilirubin to have more antioxidant capability than vitamin E, which is commonly assumed to be the most potent antioxidant in the human system. [5] This possible role of bilirubin in early protection against oxidative injury, coupled with identification of multiple neonatal mechanisms to preserve and potentiate bilirubin production, has led to speculation about an as-yet-unrecognized beneficial role for bilirubin in the human neonate.
Pathophysiology
Kernicterus results from an increased concentration of indirect, or unconjugated, bilirubin. The pathophysiology depends on the underlying condition, of which the most common are Crigler-Najjar syndrome, Gilbert syndrome, hemolytic disorders, and a reduced ability to conjugate bilirubin in infants. [1]
Bilirubin staining can be noted on autopsy of fresh specimens in the regions of the basal ganglia, hippocampus, substantia nigra, and brainstem nuclei. Such staining can occur in the absence of severe hyperbilirubinemia; in this situation, factors influencing permeability of the blood-brain barrier (eg, acidosis, infection) and the amount of unbound (versus albumin-bound) bilirubin may play a role.
Characteristic patterns of neuronal necrosis leading to the clinical findings consistent with chronic bilirubin encephalopathy are also essential in the pathophysiology of this entity. Bilirubin staining of the brain without accompanying neuronal necrosis can be observed in babies who did not demonstrate clinical signs of bilirubin encephalopathy but who succumbed from other causes. This staining is thought to be a secondary phenomenon, dissimilar from the staining associated with kernicterus.
Improved brain imaging modalities, such as magnetic resonance imaging (MRI) and ultrasonography, may be emerging as instrumental tools to help clarify the complex picture of kernicterus in contrast with asymptomatic bilirubin staining of brain tissues. Bilirubin staining has been suggested to be visualized on MRI as an increased signal in the posteromedial aspect of the globus pallidus. Despite its theoretical value, however, efforts to use cranial imaging in the clinical setting have remained unsatisfying. A 2008 case series by Gkoltsiou et al reported the inexplicable conclusion that, while all children with severe cerebral palsy and a history of hyperbilirubinemia had abnormal central grey matter on later scans, the characteristic central grey matter MRI features of kernicterus were not seen in early scans. [6]
Etiology
Familiarity with bilirubin metabolism leads to an understanding of the factors leading to an increased risk of kernicterus (see image below). Bilirubin is produced during the catabolism of the heme component of red blood cells (RBCs). Red cell destruction is usually increased in the immediate neonatal period; it can be pathologically elevated in the presence of immune-mediated or nonimmune-mediated hemolytic disease. The first enzyme in the catabolic cascade leading to bilirubin is heme oxygenase. A constitutive form and an inducible form are recognized and are induced by physiologic stressors. The creation of bilirubin, a potentially toxic water-insoluble compound, from biliverdin, a nontoxic water-soluble substance, consumes energy.
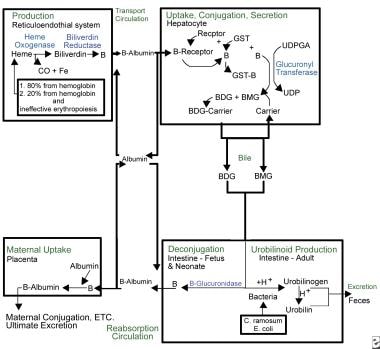
Because of its lipophilic nature, bilirubin must be bound to albumin to travel through the blood stream. In this state, it is not free to cross the blood-brain barrier and cause kernicterus. The albumin-bilirubin complex is carried to the liver, where bilirubin enters the hepatocyte for further metabolism. Once in the liver, bilirubin is conjugated via the action of uridine diphosphate glucuronyl transferase (UDPGT), [7] an enzyme not fully functional until 3-4 months of life.
Conjugated bilirubin is excreted into the intestinal tract via the biliary system. Beta-glucuronidase, present in the intestinal lumen of human neonates, deconjugates the conjugated bilirubin, allowing it to be reabsorbed across the intestinal lipid cell membranes back into the blood stream where it must be re-bound to albumin to repeat the cycle. This process, called enterohepatic recirculation, is a unique neonatal phenomenon and contributes significantly to physiologic jaundice. Feeding and excretion of meconium and stool interrupt the enterohepatic recirculation.
Among infants reported in the US kernicterus registry, 56% had abnormalities known to increase the bilirubin concentration in the blood. [8] Severe hemolytic processes were identified in 25 of 122 babies (20.5%); glucose-6-phosphate dehydrogenase deficiency was diagnosed in 26 babies (21.3%), birth trauma identified in 18 patients (15%), and other causes such as galactosemia, Crigler-Najjar syndrome, and sepsis were diagnosed in 8 babies (7%). In 53 of 122 infants (43.4%), no etiology for the severe hyperbilirubinemia was discovered.
Increased bilirubin production
Most of the circulating bilirubin in the neonate arises from destruction of circulating RBCs. Neonates produce bilirubin at more than double the daily rate of the average adult, primarily because of the larger circulating volume of RBCs and their shorter life span. Any event resulting in increased serum bilirubin load puts the infant at risk for hyperbilirubinemia.
Polycythemia
Prenatal factors, such as maternal smoking, maternal illness, placental insufficiency, and gestation at high altitude, can result in neonatal polycythemia. Obstetric factors, such as delayed clamping of the cord, stripping the cord, or holding the baby below the level of the introitus for a prolonged period, can result in increased RBC mass in the baby. This is particularly true for babies born in the absence of a trained birth attendant.
Hemolysis
Immune hemolytic disease, most often Rh isoimmunization (erythroblastosis fetalis), is the prototype etiology for kernicterus.
ABO isoimmunization, as well as minor blood group antigens, can also cause hemolytic disease in the newborn, usually of moderate severity. Infants born to mothers of blood type O negative are at greatest risk, with one series of 249 infants with severe hyperbilirubinemia reporting an odds ratio of 48.6 for infants with Rh incompatibility. [9]
Abnormalities of the red cell itself can also predispose to hemolysis. These can be grouped into membrane defects, such as hereditary spherocytosis and elliptocytosis; enzyme defects, such as glucose-6-phosphate dehydrogenase deficiency and pyruvate kinase deficiency; and hemoglobinopathies, such as alpha and beta thalassemias.
Sickle cell disease does not typically cause hemolytic disease in the neonatal period.
Extravasated blood
Significant areas of bruising, such as severe cephalohematoma, subgaleal hemorrhage or peripheral ecchymoses from birth trauma, can result in an increased bilirubin load in the serum as the blood collection resolves. Internal areas of hemorrhage, such as pulmonary or intraventricular bleeds, can also be a significant occult source of serum bilirubin.
Enzyme induction
As mentioned above, heme-oxygenase-one (HO-1) is the inducible form of the first enzyme involved in the creation of bilirubin. This enzyme is activated by physiologic stressors, such as hypothermia, acidosis, hypoxia, and infection (odds ratio 20.6 in sepsis). [9]
Epidemiologic factors
East Asian and Native American babies produce bilirubin at higher rates than do White infants; Black infants have lower production rates than do infants of other racial groups. Male infants have higher serum bilirubin levels than females. Hyperbilirubinemia also runs in families; the etiology is unclear but may relate to genetically increased levels of beta-glucuronidase in the infant, in the mother's breast milk, or both (if the infant is breastfed).
Decreased elimination
Even with normal bilirubin production, abnormalities in transport, excretion, or both can result in an increased level of free bilirubin in the serum.
Albumin binding
Because of its lipophilic nature, bilirubin must be bound to carrier protein to be transported in the aqueous environment of the serum. Albumin has one primary high-affinity binding site for bilirubin and two lower-affinity sites. At physiologic pH, the amount of free bilirubin (eg, bilirubin not bound to albumin) is very low. This is important because only free bilirubin is available to cross the blood-brain barrier and cause neurotoxicity. Decreased albumin binding capacity, decreased albumin binding affinity, or both can serve to increase the amount of free serum bilirubin. Binding affinity is lower in neonates than in older infants and is lower still in premature and sick infants than in healthy term ones.
Some authors advocate including measures of unbound (ie, free) bilirubin when assessing the risk of bilirubin neurotoxicity, [10] in part because some studies have shown a closer association between the unbound bilirubin concentration and auditory abnormalities than those seen with total serum bilirubin, although identifying the neurotoxic unbound bilirubin concentration threshold remains elusive. [11]
Decreased binding capacity can occur in hypoalbuminemia or if the binding sites are filled with other anions. Whether parenterally administered lipid can displace bilirubin from its albumin-binding site is controversial. If faced with dangerously high levels of serum bilirubin, restricting lipid administration to less than maximal levels may be prudent. Drugs, such as sulfisoxazole and ceftriaxone, can also compete for bilirubin-binding sites on the albumin molecule and must be used with caution or avoided in the neonatal period.
Hepatic uptake and conjugation
Albumin carries bilirubin to the liver, where it is incorporated into the hepatocyte by an acceptor protein called ligandin. Hepatic levels of ligandin do not reach adult values until around age 5 days, but they can be induced by administration of phenobarbital.
Once inside the hepatocyte, bilirubin is conjugated to a sugar moiety, glucuronic acid, via the enzyme UDPGT. Inherent neonatal deficiency of this enzyme is the principal etiology of physiologic jaundice. For the first 10 days of life, UDPGT is present at levels about 0.1% of adult values, and hyperbilirubinemia appears to be the primary stimulus to enzyme production.
Beyond physiologic jaundice, congenital inherited defects in UDPGT cause pathologic hyperbilirubinemia of varying severity. Crigler-Najjar syndrome type I is the virtual absence of UDPGT and is characterized by profound refractory hyperbilirubinemia with the ongoing risk of kernicterus at any point during an individual's lifespan. Currently, liver transplantation is the only definitive therapy, although experimental therapies are under investigation. Patients with Crigler-Najjar syndrome type II (ie, Arias syndrome) have a similar clinical presentation as patients with type I. However, patients with type II dramatically respond to therapy with phenobarbital, which is how the diagnosis is made.
Gilbert syndrome is characterized by a benign chronic indirect hyperbilirubinemia without evidence of liver disease or abnormality. The genetic basis for this syndrome was identified as an amplified triplet repeat in the coding gene for UDPGT, and investigations are continuing to clarify the possible role of Gilbert syndrome in infants with neonatal hyperbilirubinemia.
Excretion
Once conjugated, water-soluble bilirubin is excreted in an energy-dependent manner into the bile canaliculi for ultimate delivery into the small intestine. Disruption in this system or obstruction in the biliary system results in accumulation of conjugated bilirubin in the serum, identified by an elevation in the direct fraction of total bilirubin. Direct hyperbilirubinemia in the neonate (defined as a direct fraction greater than one third of total bilirubin) is always pathologic, and an etiology must be pursued.
In the small intestine, conjugated bilirubin cannot be reabsorbed. Intestinal florae convert it into urobilinogen, which is excreted. In the neonate, the paucity of colonic bacteria impedes this conversion. Furthermore, the neonatal gut (but not that of the adult) produces beta-glucuronidase, an enzyme that acts upon conjugated bilirubin, releasing free bilirubin for potential absorption across the intestinal cell lipid membrane into the blood stream. Breast milk also contains beta-glucuronidase, and breast milk feedings increase the level of this enzyme in the neonatal intestine. Combined with slow intestinal motility in the first few days of life, the above factors result in what is called enterohepatic recirculation of bilirubin back into the blood stream.
Systemic factors
Galactosemia
Patients with this rare inborn error of metabolism may primarily present with hyperbilirubinemia, although the direct fraction typically increases during the second week of life. The baby may manifest other characteristic signs, such as hepatomegaly, poor feeding, or lethargy. Urine for reducing substances, but not glucose, is diagnostic. Many state newborn metabolic screens include a test for this disorder.
Hypothyroidism
Although the etiology is unclear, prolonged indirect hyperbilirubinemia is one of the typical features of congenital hypothyroidism, and this diagnosis must be ruled out in any baby with hyperbilirubinemia persisting after age 2-3 weeks. Most state metabolic screens include an assay of thyroid function, although false-negative results and delayed receipt of results may necessitate individual testing in symptomatic infants.
Drugs
Maternal administration of oxytocin, diazepam, or promethazine may result in increased serum bilirubin in the infant. Similarly, neonatal administration of pancuronium and chloral hydrate increases bilirubin levels. Additionally, some drugs, such as sulfonamides and some penicillins, can displace bilirubin from its albumin-binding site, effectively increasing the serum concentration of free bilirubin available to cross the blood-brain barrier.
Acidosis
Systemic acidosis decreases the binding affinity of albumin for bilirubin, resulting in increased levels of free bilirubin in the blood stream. Ready availability of protons promotes the formation of bilirubin acid (free bilirubin anion plus 2 hydrogen ions); that moiety demonstrates increased binding and transport into neural cell membranes.
Disrupted blood-brain barrier
The neonatal blood-brain barrier is more permeable to substances than is the adult's. Administration of hyperosmolar substances, hypercarbia, asphyxia, infection (particularly meningitis), and impaired autoregulation with variations in blood pressure all may weaken capillary tight junctions, increasing capillary permeability. This, in turn, might lower the concentration at which bilirubin is toxic to the CNS.
Breast milk feedings
The well-described physiologic jaundice observed in the first few days of life, particularly in the breastfed infant, is called breastfeeding jaundice. Breastfeeding jaundice is thought to result from multiple mechanisms, described above, which promote production and inhibit excretion of bilirubin, as well as from insufficient milk intake because of reduced mammary gland milk production in the first few days postpartum. Breastfeeding jaundice should be distinguished from breast milk jaundice.
Some breastfed infants, although clinically thriving, continue to manifest an indirect hyperbilirubinemia of unidentifiable etiology for several months. If this is witnessed in a breastfed infant, the exclusion diagnosis of breast milk jaundice may be made. Such hyperbilirubinemia is thought to be caused by persistently high levels of as-yet-unidentified components in some women's breast milk, which result in persistence of the infant's hyperbilirubinemia. One clue may be a history of similar hyperbilirubinemia in other breastfed siblings. This entity is benign.
Epidemiology
In general, kernicterus affects Black and South Asian children. [1]
United States data
The exact incidence of kernicterus is unknown. [1] However, Asian, Hispanic, Native American, and Eskimo infants produce higher levels of bilirubin than White infants, whereas bilirubin production is lower in Black infants. [1]
A pilot kernicterus registry monitoring the cases of babies with kernicterus in the United States who have been voluntarily reported shows 125 babies with chronic kernicterus enrolled in the registry from 1984-2002. [12, 13] All but four babies reported in the registry had been discharged from the hospital fewer than 72 hours after birth (97%). Five babies were born at home (4%). No sequelae were identified in nine of 115 infants, and one was lost to follow-up.
International data
In Denmark, eight cases of kernicterus were reported from 1994-2002, whereas no cases had been reported for the preceding 20 years. [14] Following this report, from 2002-2005, a more vigilant approach was taken to the management of newborn jaundice, and no more cases have been reported in Denmark. [15] These combined data result in an overall incidence of kernicterus in Denmark of 1.1 in 100,000 live births from 1994-2005.
In a more recent Danish study (2020) that evaluated data from 408 infants (gestational age ≥35 weeks' gestation) between 2000 and 2015, blood type ABO isohemolytic disease was the most common etiology for extreme neonatal hyperbilirubinemia and kernicterus spectrum disorder. [16] Extreme neonatal hyperbilirubinemia occurred at an incidence of 42 per 100,000 live births (with a falling incidence in 2005-2015), and the incidence of kernicterus spectrum disorder was 1.2 per 100,000 live births(12 of 408 infants). [16]
In June 2003, The Quarterly Bulletin of the Royal College of Paediatrics and Child Health announced the commencement of a surveillance program of cases of severe neonatal hyperbilirubinemia following anecdotal reports throughout Britain and Ireland of increasing observation of kernicterus. [17] A 2004 UK surveillance study reported kernicterus occurring at a rate of 1 in 100,000 live births.
A Canadian survey published in 2004 assessed the frequency of extreme hyperbilirubinemia (serum bilirubin >427 μmol/L or >25 mg/dL) as 1:2,840 live births, of which 13 (2 in 100,000) had abnormal neurological outcomes at the time of discharge. [18]
Using these data, the risk of developing kernicterus in infants manifesting extreme hyperbilirubinemia (>25 mg/dL) can be estimated across populations. In Canada, this risk calculates to 1 in 17.6 infants, whereas in Denmark, the population risk is estimated as 1 in 16.2. [19] When the threshold of extreme hyperbilirubinemia is increased to >30 mg/dL (>513 μmol/L), the risk of developing kernicterus increases to 1 in 5.5-7 live births, depending on the reports. It should be noted that kernicterus also occurs in infants in whom bilirubin levels remained < 25 mg/dL, and the population risk of this occurrence remains unknown.
Hispanic and Asian populations appear to have a greater propensity to develop hyperbilirubinemia, although the underlying explanation for this observation remains elusive. Genetic variants, such as Gilbert disease or G6PD deficiency that occur in sequestered populations, result in geographic and/or ethnic differences in the risk and frequency of kernicterus.
Race-related demographics
Among infants reported in the US kernicterus registry, 58% were White. [13] Asian and Hispanic babies born either in their native countries or in the United States and Native American and Eskimo infants have higher production levels of bilirubin than White infants. Black infants have lower production levels (see image below). The reasons for these racial differences have not been fully elucidated.
 Kernicterus. Typical patterns of total serum bilirubin levels in neonates of different racial origins. Used with the permission of the Academy of Pediatrics.
Kernicterus. Typical patterns of total serum bilirubin levels in neonates of different racial origins. Used with the permission of the Academy of Pediatrics.
Sex-related demographics
Male infants have consistently higher levels of serum bilirubin than do female infants. [1] Among infants reported in the US kernicterus registry, 67% of the patients were male. [13]
Age-related demographics
Acute bilirubin toxicity appears to occur in the first few days of life of the term infant. An estimated 60% of term and 80% of preterm infants develop jaundice in the first week of life. [20] Preterm infants may be at risk of toxicity for slightly longer than a few days. If injury has occurred, the first phase of acute bilirubin encephalopathy appears within the first week of life.
The pilot kernicterus registry data show that, of 122 infants (all >35 weeks' gestational age at birth), symptoms became apparent in 13 babies (10.6%) aged 3.5 days or younger and in 66 babies (54%) aged 4-7 days. [8] In 36 babies (29.5%), symptoms did not appear until after the first week of life. Most of these babies (76%) were term infants (at least 37 completed weeks' gestation), and no infant was younger than 35 weeks' estimated gestational age. (See the Gestational Age from Estimated Date of Delivery calculator.)
Patient Education
To facilitate the provision of appropriate evaluation and follow-up for babies without recognized risk factors, the American Academy of Pediatrics (AAP) has published an hour-of-age-specific guideline that correlates total serum bilirubin levels with degree of risk and recommendations for follow-up. [21]
The AAP recommends professional medical evaluation in 2-3 days for babies who are discharged from the hospital fewer than 48 hours after birth. [21] Babies discharged fewer than 72 hours after birth may also be at risk, and they should be closely monitored as well. Other risk factors warranting additional vigilance may include unexplained family history of neonatal hyperbilirubinemia, near-term gestation, low birth weight, excessive bruising or hematomata, and ethnicity at risk for exaggerated hyperbilirubinemia.
Parents should be informed of the importance of keeping these appointments, as well as be familiarized with the symptoms of poor feeding in breastfed babies and how to seek help.
-
Kernicterus. Typical patterns of total serum bilirubin levels in neonates of different racial origins. Used with the permission of the Academy of Pediatrics.
-
Kernicterus. Overview of bilirubin metabolism.
-
Kernicterus. Hour-specific nomogram for total serum bilirubin and attendant risk of subsequent severe disease in term and preterm infants. Used with the permission of the Academy of Pediatrics.
-
Kernicterus. Magnetic resonance image of a 21-month-old with kernicterus. The area of abnormality is the symmetric high-intensity signal in the area of the globus pallidus (arrows). Courtesy of MJ Maisels.
-
Kernicterus. Neuronal changes observed in kernicterus. Courtesy of JJ Volpe.




