Practice Essentials
Hypomelanosis of Ito (HI) syndrome is characterized by the presence of whirled hypochromic skin lesions (see the image below) often associated with systemic manifestations.
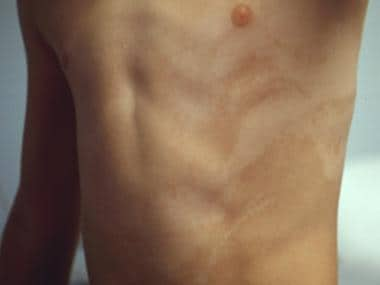 Hypomelanosis of Ito on the torso.
Hypomelanosis of Ito on the torso.
Pathophysiology
The pathogenesis of hypomelanosis of Ito syndrome is strongly linked to its genetics. See Pathophysiology.
Patient education
Psychomotor treatment may be desirable to promote the emotional-relational component of an affected child's development. [1] Parents need to be educated about this disorder, its genetics, and ways of encouraging an affected child to function as favorably as possible.
Signs and symptoms
See Physical Examination.
Diagnostics
Also see Imaging Studies and Histologic Findings.
In patients with seizures, an EEG is indicated to show focal discharges and slowing.
Most patients with cardiac anomalies require an ECG.
Perform a slit-lamp examination in patients with ophthalmologic abnormalities.
Biopsies of affected and nonaffected skin are sometimes indicated.
In select patients with cardiac anomalies, cardiac catheterization is recommended for proper diagnosis.
Chromosomal analysis
Blood karyotyping is indicated, especially when systemic abnormalities are present.
Fibroblast karyotyping (sampling the dark and light skin) can reveal mosaicism but is not mandatory for diagnosis.
Exome sequencing, using patients' blood and skin samples, may be a first choice for detecting causative genetic variants of pigmentary mosaicism. [2]
Treatment
Also see Surgical Care and Consultations.
No specific treatment is available for hypomelanosis of Ito (HI).
Treat seizures depending on the specific seizure type and epileptic syndrome (see Status Epilepticus, Complex Partial Seizures).
Partial seizures may respond to the usual anticonvulsant medications, such as carbamazepine, phenytoin, lamotrigine, gabapentin, and topiramate.
Infantile spasms should be treated accordingly with adrenocorticotropic hormone (ACTH), vigabatrin, valproic acid, or topiramate.
Approximately 30% of patients with seizures do not respond to anticonvulsant medications; therefore, these patients may need an appropriate evaluation to verify if they are good candidates for resective epilepsy surgery, ketogenic diet, or vagal nerve stimulation. In these patients, perform a prolonged video-EEG to document the zone of ictal onset.
Always offer the patient and parents genetic consultation.
Traditional depigmenting agents (eg, hydroquinone, corticosteroids, kojic acid) may be used. [3] In addition, active compounds isolated from plants (eg, arbutin, aloesin, gentisic acid, flavonoids, hesperidin, licorice, niacinamide, yeast derivatives, polyphenols) may inhibit melanogenesis without melanocytotoxicity and merit further evaluation.
No dietary restrictions are indicated. Patients with seizures who are unresponsive to anticonvulsant medication may benefit from a high-fat, low-carbohydrate diet (ie, the ketogenic diet).
Prevention
Hypomelanosis of Ito (HI) cannot be prevented, except in rare cases of familial hypomelanosis of Ito.
Because familial hypomelanosis of Ito is autosomal dominant, genetic counseling is indicated as a way to prevent new cases in the same family. Nonetheless, most cases are a de novo occurrence.
Background
Ito first introduced the syndrome 1951. [4] In 1967, Hamada et al confirmed the association between the skin lesions and systemic abnormalities, including intellectual disability. [5] Finally, Pascual-Castroviejo et al delineated the full spectrum of associated neurological abnormalities in a systematic study of the largest series published. [6]
Incontinentia pigmenti (IP) achromians is another term for this syndrome; however, because no true IP (melanin absent in the epidermis and present in the dermis) is present in the skin specimens, hypomelanosis of Ito syndrome has become the preferred name.
In 1992, Ruiz-Maldonado and associates established some diagnostic criteria for hypomelanosis of Ito syndrome. [7] Nonetheless, Ruiz-Maldonado et al's criteria link the diagnosis to the presence of systemic nondermatological (eg, CNS, skeletal) or chromosomal abnormalities. These criteria exclude patients with only dermatological manifestations. Patients with skin manifestations suggestive of hypomelanosis of Ito with and without systemic alterations have been described in the same family, demonstrating that hypomelanosis of Ito syndrome’s systemic involvement can vary. Studies that do not include systemic manifestations as diagnostic criteria for hypomelanosis of Ito reported that approximately 30-74% of patients with typical hypomelanosis of Ito skin lesions do not have nondermatological pathology.
Pathophysiology
A karyotype analysis survey was performed on 115 patients and revealed chromosomal anomalies in 60 (52%). [8] Many patients have a chromosomal mosaic pattern, often leading to the generation of 2 cell lineages, which produce patterns of hypopigmented and hyperpigmented skin. X-chromosome alterations are not unusual in hypomelanosis of Ito syndrome, and recent evidence points to X-chromosome inactivation, activation, and mosaicism as the main causes of these different patterns of cell behavior in the skin.
The nevus of Ito, like Nevus of Ota, is a hyperpigmentary dermal melanocytosis developing as a consequence of disturbances or failures during migration of melanocytes from the neural crest towards the epidermis. [9]
Perhaps this can also be found in other tissues, such as the fundus (tessellated or radial pigmentation of the fundi), iris (hypopigmentation), and the brain (areas with abnormal cell morphology and neuroblast migration side by side with normal patterns). Karyotyping the blood cells may not be diagnostic; a skin biopsy for fibroblasts may be necessary to detect the hypomelanosis of Ito–related chromosomal anomalies.
Despite recent advances, the genetic substrate for hypomelanosis of Ito syndrome is far from homogenous and is not completely understood. A wide range of chromosomal abnormalities may be observed, including balanced X-autosome translocations, supernumerary X-chromosome ring fragment, ring chromosome 10, mosaic triploidy, mosaic trisomies (8, 13, 14, 18, 22), mosaic translocations, and mosaic deletions. Autosomal deletions and duplications may involve chromosomes 7, 12, 13, 14, 15, and 18. The pattern of chromosomal aberrations and the polymorphic nature of this disease have led some to believe that hypomelanosis of Ito syndrome is a descriptive term rather than a true syndrome.
A familial form of hypomelanosis of Ito syndrome is noted; however, less than 3% of the patients have a family history of hypomelanosis of Ito–type skin lesions. Although hypomelanosis of Ito syndrome is most commonly a de novo occurrence, familial cases appear to be transmitted as an autosomal dominant trait. In one family, a 16% trisomy 2 mosaicism was identified. [10]
Approximately 10% of the patients report a family history of seizures or epilepsy, but the phenotypic expression varies; therefore, pigmentary changes may be the only clue to the genetic basis. A cutaneous ultrastructural study that shows abnormal nerve termination in close proximity to basal keratinocytes, degenerated melanocytes, premelanosomes, and Langerhans cells has suggested that this finding may be important in the pathogenesis of hypomelanosis of Ito. [11]
Etiology
Several chromosomal abnormalities have been reported in hypomelanosis of Ito (see Pathophysiology), but the etiology remains elusive.
Epidemiology
To date, epidemiological data on this syndrome are limited. It appears to be the third most common neurocutaneous disease, second only to neurofibromatosis and tuberous sclerosis. In a pediatric neurology service in Spain, 1 in 600-700 patients referred was diagnosed with hypomelanosis of Ito syndrome. It is diagnosed in 1 of every 7805 general pediatric outpatient visits, 1 of every 790 pediatric dermatology clinic visits, and 1 of every 2983 children in a general pediatric service. [6] Approximately three fourths of the patients with typical skin lesions have systemic manifestations.
Sex
Male-to-female ratios vary. In earlier series, the male-to-female ratio was reported to be 1:2.5. However, in larger and more recent series, the male-to-female ratio was 1:1.2. The severity of systemic manifestations appears to be similar in both sexes.
Age
Data in relation to age of diagnosis are usually reported in regard to the skin manifestations of hypomelanosis of Ito syndrome. Typical skin lesions are initially demonstrated during the first year of life in as many as 70% of patients; they are noticeable at birth in 54% of patients. Rarely, lesions are not visible until mid childhood.
Prognosis
Prognosis depends on the patient’s manifestations and complications of the disease. About three fourths of patients with typical hypomelanosis of Ito skin lesions have systemic manifestations of the disease.
As many as three fourths of patients have neurological symptoms in the first decade of life. Cognitive deficit is defined as an IQ of less than 70. Association of intellectual disability and seizures suggests hypomelanosis of Ito, although the cause-effect link is difficult to prove, except in the intractable cases.
Patients with chromosomal anomalies are at risk for tumors.
-
Hypomelanosis of Ito on the torso.



