Practice Essentials
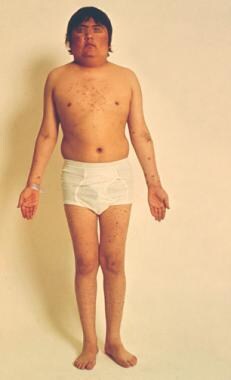
Hyperpituitarism, or primary hypersecretion of pituitary hormones, is rare in children. It typically results from a pituitary microadenoma. [1] If laboratory findings suggest pituitary hormone excess, the presence of a pituitary adenoma should be confirmed with magnetic resonance imaging (MRI). Prolactinoma is the only pituitary adenoma for which long-term medical management is first-line treatment. For patients with acromegaly and for those with Cushing disease (see the image below), the preferred treatment is surgery.
Signs and symptoms
The clinical presentation of a pituitary adenoma primarily results from the oversecreted hormone. The tumor mass itself may cause headaches, visual changes due to optic nerve compression, or hypopituitarism.
Signs and symptoms of excess prolactin include the following:
-
Headache, visual disturbance, and growth failure in prepubertal children
-
Pubertal arrest or hypogonadism (with or without galactorrhea) in female adolescents
-
Headaches, visual impairment, and pubertal arrest or growth failure in male adolescents
The following signs and symptoms are associated with excess adrenocorticotropic hormone (ACTH):
-
Generalized weight gain with concurrent growth failure (usually precedes other manifestations)
-
Hirsutism and premature adrenarche in prepubertal children
-
Pubertal arrest
-
Acne
-
Fatigue
Signs and symptoms of excess growth hormone (GH) include the following:
-
Tall stature
-
Mild to moderate obesity (common)
-
Macrocephaly, which may precede linear growth
-
Exaggerated growth of the hands and feet with thick fingers and toes
-
Coarse facial features, including frontal bossing and prognathism
-
Menstrual irregularities
-
Ectopic point of maximal impulse (PMI) caused by left ventricular hypertrophy
-
Benign tumors, such as skin tags
See Presentation for more detail.
Diagnosis
Laboratory studies
The following laboratory studies are usually included in the workup of hyperprolactinemia:
-
Serum prolactin (PRL) measurement
-
Thyrotropin-releasing hormone (TRH) stimulation test
Tests that may be included in the workup of a suspected adrenocorticotropic hormone–releasing adenoma are as follows:
-
Urinary steroid excretion measurement
-
Plasma cortisol measurement
-
Dexamethasone suppression testing
-
Plasma ACTH measurement
-
Corticotropin-releasing hormone (CRH) stimulation testing
-
Inferior petrosal sinus sampling
-
Growth hormone–releasing adenoma
The following studies are often included in the workup of a suspected growth hormone–releasing adenoma:
-
Serum insulin-like growth factor-I (IGF-I) measurement
-
Serum insulin-like growth factor-binding protein-3 (IGFBP-3) measurement
-
Oral glucose tolerance test
Imaging studies
A T1-weighted spin-echo MRI scan of the pituitary before and after administration of gadolinium is the imaging modality of choice for detecting pituitary adenomas.
See Workup for more detail.
Management
For most patients with a prolactinoma, medical management with dopamine agonists should be attempted before surgery is considered. Somatostatin analogs are highly effective for patients with growth hormone excess.
The treatment of choice for patients with Cushing disease is transsphenoidal microsurgery. Similarly, the preferred primary treatment for patients with acromegaly is surgery.
See Treatment and Medication for more detail.
Background
The most frequently encountered adenoma in children is the prolactinoma, followed by corticotropinoma and somatotropinoma. Fewer than 20 cases of thyrotropinoma in children have been reported, all with onset after age 11 years. Pediatric gonadotropinoma has not been reported.
Hypersecretion of pituitary hormones secondary to macroadenomas (see the image below) can interfere with other pituitary hormone functions, resulting in target organ hormone abnormalities (hypogonadism, hyperadrenalism, hyperthyroidism, or hypothyroidism).
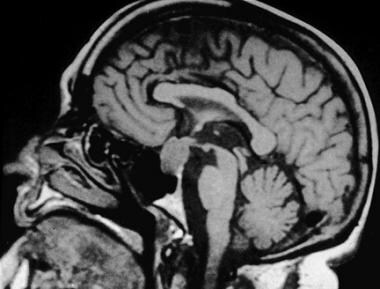
In some cases, long-standing hormonal hypersecretion is accompanied by sufficient hyperplasia of the pituitary to produce sellar enlargement.
Elevated pituitary hormone levels can also result from normal physiologic responses to abnormalities in target organs. Hyperpituitarism resulting from primary endocrine organ deficiency (eg, high circulating thyroid-stimulating hormone [TSH] levels in primary hypothyroidism due to Hashimoto thyroiditis or elevated adrenocortical hormone [ACTH] levels in primary adrenal insufficiency) quickly suppress to reference range values upon replacement of the active hormone. This form of hyperpituitarism is not accompanied by pituitary adenoma.
Rarely, ectopic tumors can secrete pituitary hormones. Neuroendocrine cells are found in the lung, gastrointestinal tract, pancreas, thyroid gland, adrenal medulla, breast, prostate, and skin and produce hormone products. Neuroendocrine tumors comprise the most common of the “ectopic” hormone syndromes. These cells can produce ACTH, growth hormone-releasing hormone (GHRH), corticotropin-releasing hormone (CRH), somatostatin (SRIH) among many other hormones.
This article focuses on the endocrine manifestations of pituitary adenomas in children.
Pathophysiology
Hypothalamic dysfunction clearly may promote tumor growth, but overwhelming evidence indicates intrinsic pituicyte genetic disruption leads to pituitary tumorigenesis. The monoclonal nature of most pituitary adenomas, confirmed by X-inactivation studies, implies their usual origin from a clonal event in a single cell. Most pituitary adenomas are functional and secrete a hormone that produces a characteristic clinical presentation. Nonfunctioning pituitary adenomas are rare in children, accounting for only 3-6% of all adenomas in 2 large series, whereas they comprise 30% of adenomas in adults. In children, disruption of growth regulation and/or sexual maturation is common, either because of hormone hypersecretion or because of manifestations caused by local compression by the tumor.
Prolactinoma
Overall, prolactinoma is the most common pituitary adenoma encountered in childhood. Most pediatric cases occur in adolescence, more commonly in females than males. Boys tend to have larger tumors and higher serum prolactin (PRL) levels than girls. Females with these tumors present with amenorrhea, and males present with gynecomastia and hypogonadism. Prolactinomas arise from acidophilic cells that are derived from the same lineage as the somatotropes and thyrotropes. Hence, PRL-secreting adenomas may also stain for and secrete growth hormone (GH) and, occasionally, TSH.
Corticotropinoma (Cushing disease)
In children, corticotropinomas are the most common adenomas observed before puberty, although they occur in people of all ages. They increase in frequency in pubescent and postpubescent children, with a female preponderance. First described by Harvey Cushing in the early 1900s, Cushing disease (see the images below) specifically refers to an adrenocorticotropic hormone (ACTH)–producing pituitary adenoma that stimulates excess cortisol secretion.
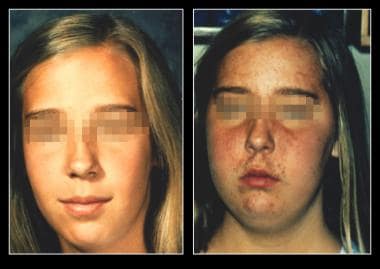 On the left is an unaffected patient aged 12 years. On the right is the same patient aged 13 years after developing Cushing disease.
On the left is an unaffected patient aged 12 years. On the right is the same patient aged 13 years after developing Cushing disease.
Adenomas that cause Cushing disease are significantly smaller than all other types of adenomas at presentation. Children have clinical courses somewhat different from adults. They most commonly present with weight gain (usually not centripetal) and growth failure. As in adults, most patients display an absence of the physiologic diurnal rhythm of plasma cortisol and ACTH with increased urinary excretion of free cortisol and 17-hydroxycorticosteroids (17-OHCS).
Somatotropinoma (gigantism)
GH-secreting adenomas are rare in childhood. Gigantism refers to GH excess in childhood when open epiphysial plates allow for excessive longitudinal growth. Most cases of gigantism result from GH-secreting pituitary adenomas or hyperplasia. Although gigantism is thought to occur as an isolated disorder, it occasionally represents one feature of other conditions (eg, multiple endocrine neoplasia [MEN] type 1, McCune-Albright syndrome [MAS], neurofibromatosis, tuberous sclerosis, Carney complex). In both European and Mexican cohorts, mutations in AIP, aryl hydrocarbon receptor-interacting protein, have been associated with early onset gigantism. [2, 3] There are also recent studies describing other genetic underpinnings of childhood growth hormone secreting adenoma. In two separate series, microduplications in Xp26.3 were found to be an important mutation causing early onset gigantism. [4, 5]
Mammosomatotrophs are the most common type of GH-secreting cells in childhood gigantism; hence, GH-secreting adenomas often stain for and secrete PRL (67% in one study). GH-secreting tumors in pediatric patients are more likely to be locally invasive or aggressive than those in adult patients. Activating mutations of the stimulatory Gs alpha (Gsa) protein have been identified in the somatotrophs of pituitary lesions in MAS and in as many as 40% of sporadic GH-secreting pituitary adenomas.
Thyrotropinoma
Very few cases of thyrotropinoma have been reported in children. These adenomas may secrete excess PRL, GH, and alpha subunit in addition to TSH. They are usually large because of their aggressive features and because their diagnosis is often delayed. The clinical presentation consists of signs and symptoms of hyperthyroidism, visual symptoms, and headaches. Biochemical features include the elevation of circulating free thyroxine (T4) and total triiodothyronine (T3) levels but inappropriately unsuppressed TSH.
Etiology
Hypothalamic dysfunction can promote tumor growth, but overwhelming evidence points to intrinsic pituicyte genetic disruption as the main underlying cause of pituitary tumorigenesis. The monoclonal nature of most pituitary adenomas, confirmed with X-inactivation studies, implies their origin from a clonal event in a single cell.
Most pituitary adenomas are functional, and clinical presentation typically depends on the particular pituitary hormone that is hypersecreted. Nonfunctioning pituitary adenomas are rare in children, accounting for only 3-6% of all adenomas in 2 large series; they comprise 30% of adenomas in adults. [6]
Epidemiology
United States data
Although less common in children than in adults, pituitary adenomas constitute 2.7% of supratentorial tumors in children and 3.6-6% of all pituitary adenomas that are surgically treated. The average annual incidence of pituitary adenomas presenting before age 20 years is estimated to be less than 0.1 per million children. [7]
Race-, sex-, and age-related demographics
Race and ethnicity have not been reported as significant contributing factors to hyperpituitarism.
In prolactinoma, the female-to-male ratio is 4.5:1. In ACTH-releasing adenoma, the female-to-male ratio is 2:1. In GH-releasing adenoma, the female-to-male ratio is 1:2. [8]
In children, ACTH-releasing adenomas are most prevalent in the youngest group and decrease in frequency with advancing age. The incidence of prolactinomas increases with age; 93% occur in children older than 12 years. GH-releasing tumors have a fairly even distribution among the various age groups. [8]
Prognosis
The prognosis for hyperfunctioning pituitary tumors in children is very good. Medical therapy or transsphenoidal surgery are preferred methods of treatment.
A retrospective review of pituitary adenomas in pediatric patients found that the rate of recurrence was higher in adenomas with an elevated proliferation index of ≥3% (20.8%) or with evidence of local invasion (18.2%). [1]
Prolactinoma
The postoperative prolactin value, obtained 1-2 days after surgery, accurately predicts outcome. Undetectable level (< 2 µg/L) predicts cure with more than 90% probability, whereas higher values within the reference range indicate incomplete removal of the adenoma. Surgery has a good outcome, with a long-term surgical cure rate approaching 82% for all prolactinomas with very low morbidity and no mortality.
Corticotropinoma
The criteria for cure of Cushing disease are undetectable plasma cortisol concentration in the morning (< 1 µg/mL) and corticotropin concentration of less than 5 pg/mL over 24 consecutive hours measured 4-7 days after surgery (at least 24 h after withdrawal of exogenous hydrocortisone or 48 h after exogenous prednisone). Initial remission rates (1-y) of 70-98% and long-term (10-y) success rates of 50-98% have been reported.
GH-secreting adenoma
The preferred primary treatment for the patient with acromegaly is surgery, with the surgical cure rate approaching 83% in the largest series. [9] Basal serum GH levels obtained immediately after surgery indicate the risk of tumor recurrence in children with GH-releasing adenomas. One series reported that immediate postoperative GH values of approximately 50 ng/mL were more likely to be associated with tumor recurrence than values near 15 ng/mL.
Morbidity/mortality
Large long-standing pituitary tumors may cause loss of vision or changes in the visual field if they impinge on the optic nerve. Transsphenoidal pituitary surgery has emerged as the treatment of choice for ACTH-secreting and GH-secreting adenomas. Transsphenoidal surgery is indicated for prolactinomas that do not respond to medical therapy. Transsphenoidal surgery is associated with remarkably little morbidity and near zero mortality. A permanent loss of pituitary function occurs infrequently. The incidence of postoperative hypopituitarism is about 3% in patients with microadenomas and slightly increases with the invasiveness of the tumor.
Complications
Complications of hyperpituitarism are dependent on the hormone being overproduced.
Complications of treatment
The incidence of post-operative hypopituitarism is about 3% in patients with microadenomas and increases slightly with the invasiveness of the tumor.
Parasellar radiotherapy can lead to panhypopituitarism, optic nerve and optic chiasm injury, delayed radiation injury of the brain, or increased risk of a second brain tumor and epilation (loss of facial or scalp hair).
-
Pituitary macroadenoma.
-
A 16-year-old boy with Cushing disease.
-
On the left is an unaffected patient aged 12 years. On the right is the same patient aged 13 years after developing Cushing disease.





