Background
Cushing syndrome (CS) takes its name from Harvey Cushing, who, in 1912, was one of the first physicians to report a patient affected with excessive glucocorticoid. [1] More than 99% of cases of Cushing syndrome are due to administration of excessive amounts of glucocorticoid. This article discusses issues relating to both endogenous and exogenous glucocorticoid excess, with emphasis on the safest possible therapeutic use of glucocorticoids.
Although distinguishing endogenous from exogenous Cushing syndrome is usually straightforward, the investigation and differentiation of Cushing syndrome from other causes of hypercortisolism require a sound understanding of the physiology of the hypothalamic-pituitary-adrenal (HPA) axis. See the images below.
Pathophysiology
Glucocorticoid synthesis and release is strictly regulated by the pituitary and hypothalamus by negative feedback and, to a lesser extent, by catecholamines from the adrenal medulla and neural inputs from the autonomic system. In addition to the glucocorticoid effects that cortisol has because of binding to the glucocorticoid receptor (GR), cortisol can also bind to and activate the mineralocorticoid receptor (MR). When cortisol binds to the kidney, MR is physiologically inhibited by conversion of cortisol to its inactive metabolite cortisone by the enzyme 11beta-hydroxy-steroid dehydrogenase (11beta-OHSD2), which co-localizes with the MR.
The basal daily rate of cortisol secretion is approximately 6-8 mg/m2 body surface area, although this can increase as much as 10-fold in response to acute severe stress. Physiological replacement of cortisol requires higher doses of 10-15 mg/m2 because the oral bioavailability is 50-60%. Other natural and synthetic glucocorticoids are noted, all of which have different relative potencies as glucocorticoids and mineralocorticoids because of their differing structures and affinities for the GR and MR, as well as for 11beta-OHSD2. Table 1 summarizes the relative potencies and half-lives of main steroid hormones.
The glucocorticoid receptor is an intracellular protein that, in its ligand-bound form, acts as a nuclear transcription factor to regulate the expression of a diverse array of genes in many areas of the body. Factors that influence the spectrum of adverse effects observed in hypercortisolemic individuals include duration of treatment, potency of the steroid, dose and route of administration, and the site and rate of metabolism and clearance.
Since the late 1940s, when glucocorticoids first came into use for their anti-inflammatory and immunomodulatory effects, much work has been conducted by science and industry to maximize their beneficial effects while minimizing their adverse effects. Thus, many synthetic compounds with glucocorticoid activity have been manufactured and tested.
Alterations of the basic steroid nucleus and its side groups give rise to the pharmacologic differences between these chemicals. Such changes may affect the bioavailability of these steroid compounds, including their GI absorption; parenteral distribution; plasma half-life; their metabolism in the liver, fat, or target tissues; and their ability to interact with the GR and MR and modulate the transcription of glucocorticoid-responsive genes. In addition, structural modifications can diminish the natural cross-reactivity of glucocorticoids with the MR, eliminating their undesirable salt-retaining activity. Other modifications enhance their water solubility for parenteral administration or reduce their water solubility to enhance topical potency.
Most synthetic glucocorticoids (eg, methylprednisolone, dexamethasone) are minimally bound to cortisol-binding globulin and circulate freely, or they are weakly bound to albumin. A relatively constant percentage of synthetic glucocorticoids is bound to plasma proteins, and, because this percentage is concentration independent, the rate of metabolic clearance remains constant for synthetic glucocorticoids, regardless of dose. Table 1 shows the relative glucocorticoid and mineralocorticoid potencies of different, commonly used systemic glucocorticoids and their approximate plasma and biologic effect half-lives.
Glucocorticoid activity has been defined mostly in rat bioassays, which may not always reflect human responses, particularly the growth-suppressing properties of synthetic glucocorticoids, which have been markedly underestimated. Glucocorticoids can be categorized as short, intermediate, or long acting, based on their biologic effective half-life, which is defined as the duration of corticotropin (ACTH) suppression after a single dose of the compound.
Table 1. Glucocorticoid Equivalencies [2] (Open Table in a new window)
Type |
Drug |
Dose |
Relative Glucocorticoid Potency |
Relative Mineralocorticoid Potency |
Plasma Half-Life (mg) |
Biologic Half-Life (h) |
Short-acting |
Cortisol |
20 |
1.0 |
2 |
90 |
8-12 |
Hydrocortisone‡ |
25 |
0.8 |
2 |
80-118 |
8-12 |
|
Intermediate-acting |
Prednisone |
5 |
4 |
1 |
60 |
18-36 |
Prednisolone |
5 |
4 |
1 |
115-200 |
18-36 |
|
Triamcinolone |
4 |
5 |
0 |
30 |
18-36 |
|
Methylprednisolone |
4 |
5 |
0 |
180 |
18-36 |
|
Long-acting |
Dexamethasone |
0.5 |
25-50 |
0 |
200 |
36-54 |
Betamethasone |
0.6 |
25-50 |
0 |
300 |
36-54 |
|
Mineralocorticoid |
Aldosterone |
0.3 |
0 |
300 |
15-20 |
8-12 |
Fludrocortisone |
2 |
15 |
150 |
200 |
18-36 |
|
Desoxycorticosterone acetate |
0 |
0 |
20 |
70 |
… |
Endogenous Cushing syndrome
Cushing syndrome can be divided into ACTH-dependent and ACTH-independent forms. The proportion of adrenal and pituitary disease varies in different regions; however, in Western countries, 90-95% of cases of Cushing syndrome in children older than 5 years are ACTH-dependent, and 90-95% of those cases are due to Cushing disease caused by an ACTH-secreting pituitary adenoma. Tumors that ectopically secrete ACTH are rare, and tumors that secrete corticotropin-releasing hormone (CRH) are extremely rare, together accounting for fewer than 5% of cases of Cushing syndrome.
In children younger than 5 years, the proportion of ACTH-independent cases of Cushing syndrome approaches 50%. Such cases are due to a combination of congenital disorders of the adrenal cortex and adrenocortical neoplasms that result in autonomous overproduction of cortisol and other adrenal cortical hormones (summarized below). All children in this age group who have been proven to have ACTH-independent Cushing syndrome require adrenalectomy because of the significant incidence of malignancy in this age group.
Pathophysiology of ACTH-dependent Cushing syndrome
Relative frequency is as follows:
-
Age younger than 5 years - 50% of Cushing syndrome cases
-
Age older than 5 years - 80-90% of Cushing syndrome cases
ACTH-producing pituitary adenoma (corticotropinoma) represents 80-90% of ACTH-dependent Cushing syndrome cases in people of all ages. It is usually a microadenoma and may invade the cavernous sinus. It is associated with a risk of Nelson syndrome after bilateral adrenalectomy.
Ectopic ACTH production is very rare in children. Ectopic ACTH production is from carcinoid tumors (bronchial tumors most frequent, although may also be in GI tract), ACTH-producing pancreatic islet cell tumors (especially multiple endocrine neoplasia type 1 [MEN1]), pheochromocytoma, ganglioneuroma or other neuroendocrine tumor.
Ectopic CRH production is extremely rare.
Pathophysiology of ACTH-independent Cushing syndrome
Frequency is as follows:
-
Age younger than 5 years - 50% of Cushing syndrome cases
-
Age older than 5 years - 10-20% of Cushing syndrome cases
Adrenocortical neoplasms have a risk of malignancy significant in young children.
Macronodular disease is very rare in children.
Ectopic expression of receptors on cortisol-producing cells, resulting in hypercortisolemia shown in some cases [3]
Micronodular disease may include the following:
-
Primary pigmented nodular adrenal disease (PPNAD)
-
Carney complex (See Table 2.)
McCune-Albright syndrome may be present. See the discussion of McCune-Albright syndrome in Table 2.
Etiology
Cushing syndrome can be classified as ACTH-dependent and ACTH-independent. ACTH-dependent causes can be further divided according to whether ACTH secretion arises from the pituitary or from an ectopic source. ACTH-independent causes can be divided further according to whether they are due to neoplasia or hyperplasia. Table 2 summarizes the causes of Cushing syndrome.
Exogenous Cushing syndrome occurs as the result of systemic absorption of pharmacologic doses of steroids with glucocorticoid activity. Most commonly, this results from oral or parenteral administration but may also be caused by inhaled steroids, topical steroids, and, occasionally, local steroid injections.
Table 2. Genetic Causes of Cushing Syndrome (Open Table in a new window)
Cause |
Features |
Genetics |
MEN1 |
Associated with pancreatic tumors producing gastrin, insulin, and/or ACTH that may metastasize to the liver; multigland hyperparathyroidism, pituitary tumors, lipomas, and angiofibromas |
11p13 (MIM 131100) |
Mosaic constitutively activating postzygotic GS alpha mutation that can lead to polyostotic fibrous dysplasia, pigmented skin lesions, gonadotropin-releasing hormone–independent precocious puberty, hyperthyroidism, renal phosphate wasting, and other endocrine and nonendocrine manifestations |
20q13.2 (MIM 174800) |
|
Beckwith-Wiedemann syndrome (Risk of adrenal malignancy) |
Macroglossia; visceromegaly; hyperinsulinemia; omphalocele; and risk of adrenal carcinoma, nephroblastoma, hepatoblastoma, rhabdomyosarcoma, and thoracic neuroblastoma requiring biannual sonograms |
11p13 (MIM 130650) |
Hemihypertrophy (Risk of adrenal malignancy) |
Adrenal tumors in association unilateral tissue overgrowth on ipsilateral or contralateral side Compare upper and lower limbs and look for facial asymmetry |
(MIM 235000) [4] |
Li-Fraumeni syndrome (Risk of adrenal malignancy) |
Adrenal neoplasm Personal or family history of multiple tumors (eg, lung, breast, nasopharynx, CNS, melanoma, pancreas, gonads, prostate) |
17p13.1 -TP53 gene 22q12.1 (MIM 191170; 151623) |
Primary pigmented nodular adrenal disease (PPNAD); lentigines; myxomas of the heart, skin, and breast; melanotic schwannoma; growth hormone– and prolactin-secreting pituitary adenomas; Sertoli cell tumors of the testis; multiple small hypoechoic thyroid lesions; thyroid carcinoma |
2p16 and 17q22-24 (MIM 605244; 160980) |
Epidemiology
United States data
Cushing syndrome is a rare disorder, with 90% of cases occurring during adulthood. Overall incidence is estimated to be 2 new cases per million population per year. Incidence in children is estimated at approximately 0.2 cases per million population per year.
The National Cancer Institute (NCI) estimates the incidence of adrenal cortical carcinoma as 2 cases per million population per year. Pituitary causes of Cushing disease are 5-6 times more common than adrenal causes.
Prevalence of exogenous Cushing syndrome depends on the frequency and spectrum of medical conditions requiring glucocorticoid treatment in a given population. Considerable variation in this frequency is observed in populations of different cultural and ethnic backgrounds.
International data
In certain regions of the world (eg, Japan, Brazil), adrenal tumors are more frequently observed. Whether this and other aberrations are due to a genetically determined founder effect in a small subset of the population or whether environmental factors may be acting to increase patient risk is unknown.
Sex- and age-related demographics
Endogenous Cushing syndrome of pituitary etiology is more prevalent in women than in men, with a female-to-male ratio of 9:1. Females are 8 times more likely than males to develop an ACTH-secreting pituitary adenoma and 3 times more likely to develop a cortisol-secreting adrenal tumor.
Onset of endogenous Cushing syndrome of pituitary etiology occurs primarily in the third and fourth decades of life.
Prognosis
As a result of the multiple adverse effects of chronic glucocorticoid excess, both endogenous and exogenous Cushing syndrome are associated with significant morbidity. Untreated, they are also associated with an increased risk of premature death. Specific information about the effects of glucocorticoids on different systems is summarized in Table 3.
Hypothalamic-pituitary-adrenal (HPA) axis recovery
More than 90-95% of patients have recovery of their HPA axis by 12 months after stopping treatment, with more than 50% of the remainder recovering in the following 6-12 months. Permanent adrenal insufficiency has been described, although it is rare. Early recognition and prompt treatment of the early signs of adrenal insufficiency is essential because this may be life threatening if not managed appropriately.
Growth
Growth velocity usually normalizes and weight loss occurs in children once pharmacologic doses of glucocorticoids have been reduced to physiologic levels. However, catch-up growth is frequently disappointing, with a tendency not to achieve predicted final height.
Bone density
Patients with significant osteoporosis experience some recovery in bone density, provided they have adequate calcium and vitamin D replacement and regular exercise. Bisphosphonate treatment may be needed in severe cases. Residual deficits in bone density are more likely if treatment was prolonged and occurred at a time of peak bone mass accrual. The role of prophylactic treatment with bisphosphonates is still being studied.
Metabolic disorders
Diabetes and insulin resistance resolve with cessation of therapy, although patients who become frankly diabetic when on glucocorticoids are likely to have significant preexisting insulin resistance and are at risk of developing type 2 diabetes in later life. Dyslipidemia should also improve as insulin resistance resolves, although this also depends upon premorbid lipid status.
Cushing disease
With transsphenoidal pituitary surgery, the cure rate for uncomplicated cases is approximately 95% and the recurrence rate is about 5%. If evidence of cavernous sinus invasion is noted or if repeat surgery is required, the cure rate falls significantly and the complication rate also rises.
Adrenal neoplasms
For nonmalignant adrenal neoplasms, the cure rate remains excellent. For malignant tumors, surgery offers the best chance of cure or prolongation of survival, with excision of isolated metastases in the lung or lymph nodes being primary treatment. Results of chemotherapy and radiation therapy have been disappointing, and, although disease control has been achieved, cure with these methods is uncommon so they have a more palliative role.
Patient Education
Educate patients and parents to recognize situations where an increase in glucocorticoid dosage is required. Unfortunately, the medical profession often also needs education on this issue because physicians sometimes do not appreciate the urgency of treatment in the patient who is developing signs of adrenal insufficiency.
Ensure that parents and patients understand the importance of proper technique for administering their glucocorticoid treatments (eg, the need for a spacer device with asthma, the importance of using potent steroid creams sparingly).
Children with Cushing syndrome are commonly diligent workers. Warn the family that their school performance and concentration may suffer after successful treatment and that the child may also develop psychiatric symptoms, including anxiety and depression, possibly requiring psychiatric treatment. [5]
Siblings in the same household should not receive attenuated live-virus vaccines because of the risk of causing infection in the child who is affected by Cushing syndrome.
All patients receiving glucocorticoid therapy for longer than 1-2 months should be provided a medic-alert bracelet identifying them as dependent on steroids.
-
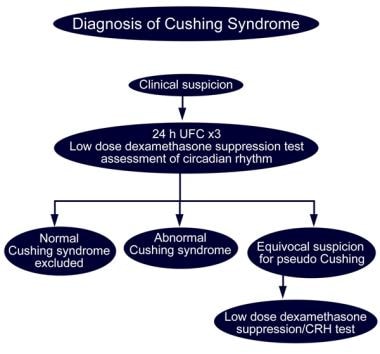
Glucocorticoid Therapy and Cushing Syndrome. Diagnosis of Cushing syndrome.
-
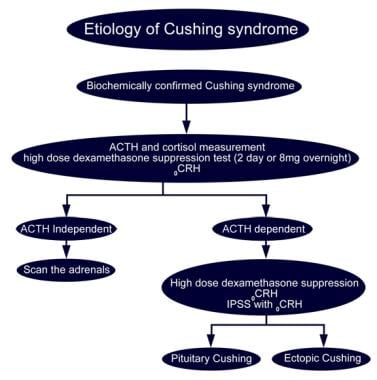
Glucocorticoid Therapy and Cushing Syndrome. Etiology of Cushing syndrome.
-
Glucocorticoid Therapy and Cushing Syndrome. Physical findings in Cushing syndrome.
Tables
Type |
Drug |
Dose |
Relative Glucocorticoid Potency |
Relative Mineralocorticoid Potency |
Plasma Half-Life (mg) |
Biologic Half-Life (h) |
Short-acting |
Cortisol |
20 |
1.0 |
2 |
90 |
8-12 |
Hydrocortisone‡ |
25 |
0.8 |
2 |
80-118 |
8-12 |
|
Intermediate-acting |
Prednisone |
5 |
4 |
1 |
60 |
18-36 |
Prednisolone |
5 |
4 |
1 |
115-200 |
18-36 |
|
Triamcinolone |
4 |
5 |
0 |
30 |
18-36 |
|
Methylprednisolone |
4 |
5 |
0 |
180 |
18-36 |
|
Long-acting |
Dexamethasone |
0.5 |
25-50 |
0 |
200 |
36-54 |
Betamethasone |
0.6 |
25-50 |
0 |
300 |
36-54 |
|
Mineralocorticoid |
Aldosterone |
0.3 |
0 |
300 |
15-20 |
8-12 |
Fludrocortisone |
2 |
15 |
150 |
200 |
18-36 |
|
Desoxycorticosterone acetate |
0 |
0 |
20 |
70 |
… |
Cause |
Features |
Genetics |
MEN1 |
Associated with pancreatic tumors producing gastrin, insulin, and/or ACTH that may metastasize to the liver; multigland hyperparathyroidism, pituitary tumors, lipomas, and angiofibromas |
11p13 (MIM 131100) |
Mosaic constitutively activating postzygotic GS alpha mutation that can lead to polyostotic fibrous dysplasia, pigmented skin lesions, gonadotropin-releasing hormone–independent precocious puberty, hyperthyroidism, renal phosphate wasting, and other endocrine and nonendocrine manifestations |
20q13.2 (MIM 174800) |
|
Beckwith-Wiedemann syndrome (Risk of adrenal malignancy) |
Macroglossia; visceromegaly; hyperinsulinemia; omphalocele; and risk of adrenal carcinoma, nephroblastoma, hepatoblastoma, rhabdomyosarcoma, and thoracic neuroblastoma requiring biannual sonograms |
11p13 (MIM 130650) |
Hemihypertrophy (Risk of adrenal malignancy) |
Adrenal tumors in association unilateral tissue overgrowth on ipsilateral or contralateral side Compare upper and lower limbs and look for facial asymmetry |
(MIM 235000) [4] |
Li-Fraumeni syndrome (Risk of adrenal malignancy) |
Adrenal neoplasm Personal or family history of multiple tumors (eg, lung, breast, nasopharynx, CNS, melanoma, pancreas, gonads, prostate) |
17p13.1 -TP53 gene 22q12.1 (MIM 191170; 151623) |
Primary pigmented nodular adrenal disease (PPNAD); lentigines; myxomas of the heart, skin, and breast; melanotic schwannoma; growth hormone– and prolactin-secreting pituitary adenomas; Sertoli cell tumors of the testis; multiple small hypoechoic thyroid lesions; thyroid carcinoma |
2p16 and 17q22-24 (MIM 605244; 160980) |
System |
Effects |
Endocrine and metabolic |
Suppression of hypothalamic-pituitary-adrenal (HPA) axis (adrenal suppression) Growth failure in children Hyperinsulinemia/insulin resistance Abnormal glucose tolerance test result/diabetes mellitus |
GI |
Gastric irritation, peptic ulcer Acute pancreatitis (rare, secondary to insulin resistance and hypertriglyceridemia) Fatty infiltration of liver (hepatomegaly, rare) |
Hemopoietic |
Leukocytosis Neutrophilia - Increased recruitment from bone marrow, demargination, and decreased migration from blood vessels Lymphopenia - Migration from blood vessels to lymphoid tissue Eosinopenia Monocytopenia |
Immune |
Suppression of delayed (type IV) hypersensitivity (important with Mantoux testing for tuberculosis) Inhibition of leukocyte and tissue macrophage migration Inhibition of cytokine secretion/action Suppression of the primary antigen response |
Musculoskeletal |
Osteoporosis, spontaneous fractures Avascular necrosis of femoral and humoral heads and other bones Myopathy (particularly of the proximal muscles [eg, unable to comb hair or climb stairs]) |
Ophthalmic |
Posterior subcapsular cataracts (more common in children) Elevated intraocular pressure/glaucoma |
CNS (neuropsychiatric disorders) |
Sleep disturbances, insomnia (particularly with long-acting glucocorticoids and nocturnal dosing) Euphoria, depression, mania, psychosis (more commonly observed in adults) Obsessive behaviors (children with hypercortisolism are often more studious) Pseudotumor cerebri (benign increase of intracranial pressure) |
Cardiovascular [6] |
Congestive heart failure in predisposed patients Dilated cardiomyopathy [8] |
Other cushingoid features |
Moon facies (broad cheeks with temporal muscle wasting) facial plethora Generalized and truncal obesity (more marked in adults) Supraclavicular fat collection Posterior cervical fat deposition (dorsocervical hump) Glucocorticoid-induced acne Thin and fragile skin, violaceous striae (more common in adults) Impotence, menstrual irregularity Decreased thyroid-stimulating hormone and triiodothyronine Hypokalemia (with very high cortisol levels or in the presence of potassium-wasting diuretics), metabolic alkalosis |








