Background
Hyperornithinemia-hyperammonemia-homocitrullinuria (HHH) syndrome, also called ornithine translocase deficiency, is a very rare inborn error of metabolism; the age at presentation and long-term prognosis widely vary among affected individuals. Growth and developmental delays, learning disabilities (especially speech delay), and periodic confusion and ataxia are typical presenting symptoms. In this syndrome, a defect in the transport of ornithine into the mitochondrial matrix significantly inhibits the urea cycle, thereby impeding nitrogen disposal. Early detection and treatment may lead to favorable outcome.
Pathophysiology
The urea cycle maintains the concentration of the toxic ammonium ion in a narrow, tolerable range despite a 10-fold variation in the dietary intake of its precursor, nitrogen. A total of 5 enzymes in 2 subcellular compartments (mitochondrial matrix and cytosol) convert ammonia into urea, which is excreted by the kidney (see image below).
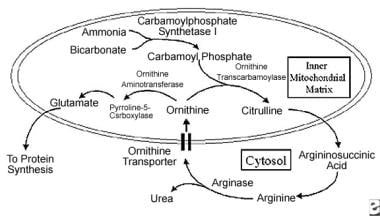 Important products and enzymes in ornithine metabolism (see text for pathway detail). Enzymes and transporters are highlighted in italics.
Important products and enzymes in ornithine metabolism (see text for pathway detail). Enzymes and transporters are highlighted in italics.
Periportal hepatocytes express these enzymes; epithelial cells of the small intestine and kidney also express these enzymes to a lesser extent, but their contribution to urea production is not significant. Urea-cycle enzyme activity is regulated by dietary protein. In part, glucagon and cyclic adenosine 3',5'-monophosphate (cAMP) regulate urea-cycle enzyme transcription.
The first 2 steps of the urea cycle occur in the mitochondrial matrix. Carbamoyl phosphate is produced from ammonia and bicarbonate by carbamoylphosphate synthetase I. This reaction is stimulated by ornithine. An inner mitochondrial membrane transporter directs ornithine to the transcarbamoylase enzyme to keep intramatrix ornithine levels low. The specifics of the liver transporter have recently been identified.
Cationic L-ornithine is electroneutrally transported into the matrix in exchange for a proton and citrulline. The inner membrane pH gradient and the availability of proton-yielding anions may affect the transport rate. As with other mitochondrial carrier family proteins, the ornithine carrier is composed of 300 amino acids that constitute 3 repeated motifs of approximately 100 amino acids each. These motifs contain 2 hydrophobic alpha-helical segments connected by an extensive hydrophilic sequence, resulting in 6 transmembrane portions of the protein.
The transporter was identified by probing a mammalian-expressed sequence tag database with 2 fungal mitochondrial ornithine carrier protein sequences. Ornithine incorporation was restored in fibroblasts derived from patients with hyperornithinemia-hyperammonemia-homocitrullinuria syndrome by transforming the fibroblasts with transporter complementary DNA (cDNA). Incorporation was traced using ornithine labeled with radioactive carbon (14C).
Following incorporation of ornithine into the mitochondrial matrix, carbamoyl phosphate and ornithine are condensed to form citrulline by ornithine transcarbamoylase. Citrulline is believed to passively diffuse across the inner mitochondrial matrix to the cytosol. The contribution of the ornithine/citrulline antiporter to citrulline transport from the mitochondria to the cytosol is not known.
The next 3 steps of the urea cycle occur in the cytosol. Argininosuccinic acid is produced from the condensation of citrulline and aspartate by a synthetase enzyme. It is then cleaved to produce fumarate and arginine by a lyase enzyme. Urea and ornithine are produced by arginase. Under normal circumstances, the ornithine produced outside the mitochondrial matrix is transported into the mitochondrial matrix, where it is reused in the urea cycle.
This transport of ornithine across the inner mitochondrial membrane is essential to the urea cycle. Ornithine can also be produced in the matrix by aminotransferase, but this enzyme is active in pericentral venous hepatocytes rather than in periportal hepatocytes.
In hyperornithinemia-hyperammonemia-homocitrullinuria syndrome, the mitochondrial ornithine transporter ORNT1 is defective. The carrier protein and gene sequence have only recently been identified; before its identification, the carrier's dysfunction was deduced biochemically because a patient with hyperornithinemia-hyperammonemia-homocitrullinuria syndrome has abnormally high ornithine levels despite normal ornithine transcarbamoylase function. Because the urea cycle cannot continue without ornithine inside the mitochondria, ammonia disposal slows, and blood ammonia levels rise. A second mitochondrial ornithine transporter, ORNT2, has been suggested and may account for a mild variation of hyperornithinemia-hyperammonemia-homocitrullinuria syndrome in French-Canadian probands. In some individuals, a gain in ORNT2 transporter function may compensate for the ORNT1 deficit.
Ornithine transcarbamoylase within the mitochondrial matrix may convert lysine to homocitrulline in the absence of ornithine, causing high blood levels of homocitrulline and homocitrullinuria. However, this theory is controversial because some studies have shown no correlation between lysine supplementation and homocitrulline levels; moreover, the role of the lysine transcarbamoylase that lies outside the inner mitochondrial membrane is not known.
Several recent studies suggest that ornithine, homocitrulline, and ammonia accumulation results in damage to the brain through increased reactive species and mitochondrial dysfunction. [1, 2, 3, 4]
Epidemiology
Frequency
International
Only about 50 cases have been reported.
Mortality/Morbidity
Neonatal death has been reported but is rare. Some patients have progressive neurologic and cognitive deterioration, whereas other patients demonstrate good function if metabolic anomalies are well controlled. Clearly, this is a very serious disorder that is potentially life-threatening and often life-shortening.
Race
Most reported cases have been in the French-Canadian population in the Quebec Province of Canada.
Sex
The male-to-female ratio is unknown.
Age
The severity ranges from minimal neurologic dysfunction in adulthood to neonatal death. Age at diagnosis also widely varies, probably, in part, because of variation in the degree of residual enzyme activity and because of the nonspecific symptoms of this disorder.
Prognosis
Growth improves with treatment.
Pregnancy is possible. A woman with hyperornithinemia-hyperammonemia-homocitrullinuria syndrome gave birth to a healthy baby on a diet that consisted of 1 g/kg of protein per day.
Some patients do not respond to diet therapy.
One case of rapidly progressive deterioration after formula feeding, leading to neonatal death, has been reported.
Older patients may develop progressive disease, but patients who have had good neurologic and cognitive function into adulthood have been reported. Abnormalities in the central and peripheral nervous systems in older patients can be detected using electrophysiologic tests (see Other Tests). The following manifestations can also occur:
-
Irritability, opposition, and aggressiveness, suggesting a conduct disorder
-
Lower IQ score (ie, in the mildly mentally retarded range)
-
Important products and enzymes in ornithine metabolism (see text for pathway detail). Enzymes and transporters are highlighted in italics.








