Background
17-Hydroxylase (17-OH) deficiency syndrome is a rare genetic disorder of steroid biosynthesis that causes decreased production of glucocorticoids and sex steroids and increased synthesis of mineralocorticoid precursors. It is a rare form of congenital adrenal hyperplasia (CAH) resulting from loss-of-function mutations involving the CYP17 gene. [1]
This syndrome is characterized by both of the following:
-
Decreased production of glucocorticoids and sex steroids, resulting in sexual infantilism in 46,XX females and ambiguous genitalia in 46,XY males
-
Increased synthesis of mineralocorticoid precursors, resulting in varying degrees of hypertension and hypokalemia
Patients are usually diagnosed with this condition during an evaluation of delayed puberty, absent secondary sexual characteristics, or primary amenorrhea. Although patients with 17-hydroxylase deficiency are cortisol deficient, they do not typically have adrenal insufficiency or experience adrenal crises. Precursor hormones such as corticosterone are elevated, have glucocorticoid activity, and are adequate to prevent adrenal insufficiency.
Exogenous glucocorticoid therapy is the treatment of choice and suppresses adrenocorticotropic hormone (ACTH) secretion, decreases 11-DOC and corticosterone levels, and normalizes serum potassium and blood pressure. Some patients may have persistent hypertension and require additional antihypertension therapy. Prognosis is generally good-to-excellent with adequate glucocorticoid therapy and monitoring.
At puberty, patients require sex steroid replacement for development of secondary sexual characteristics as well as cyclic menstrual bleeding in 46,XX females.
For patient education resources, see the Women's Health Center, as well as Amenorrhea.
Pathophysiology
Anatomically, the adrenal gland can be divided into the following three zones:
-
Zona glomerulosa, which produces predominately mineralocorticoid
-
Zona fasciculata, which produces predominately glucocorticoid
-
Zona reticularis, which produces predominately androgens
For convenience, think of the zona glomerulosa as the first endocrine organ and the zonae fasciculata and reticularis collectively as a second separate endocrine organ, as distinguished by distinct control systems.
Aldosterone (mineralocorticoid) synthesis and secretion is regulated via the renin-angiotensin system, which is responsive to the electrolyte balance state and plasma volume. Aldosterone secretion is also directly stimulated by high serum potassium concentrations. In contrast, cortisol synthesis and secretion is regulated by adrenocorticotropic hormone (ACTH), which stimulates the enzyme P-450scc (20, 22 desmolase) with subsequent increased production of all adrenal steroids in both the zona fasciculata and zona reticularis.
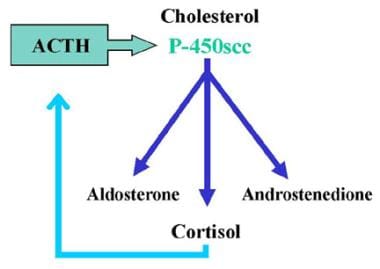 17-Hydroxylase Deficiency Syndrome. Normal adrenal steroid biosynthesis results in three products: mineralocorticoids (aldosterone), glucocorticoids (cortisol), and androgens (androstenedione). Cortisol production is regulated by feedback with adrenocorticotropic hormone (ACTH). ACTH stimulates the enzyme P-450scc (20,22 desmolase), with subsequently increased production of all adrenal steroids.
17-Hydroxylase Deficiency Syndrome. Normal adrenal steroid biosynthesis results in three products: mineralocorticoids (aldosterone), glucocorticoids (cortisol), and androgens (androstenedione). Cortisol production is regulated by feedback with adrenocorticotropic hormone (ACTH). ACTH stimulates the enzyme P-450scc (20,22 desmolase), with subsequently increased production of all adrenal steroids.
Congenital adrenal hyperplasia (CAH) is a family of autosomal recessive disorders of adrenal steroid biosynthesis in which one of the enzymes necessary for cortisol production has deficient activity. Decreased serum cortisol levels stimulate ACTH release via negative feedback. The adrenal glands undergo hypertrophy, apparently due to ACTH-stimulated production of insulinlike growth factor-2 (IGF-2). Increased ACTH secretion also results in overproduction of both the adrenal steroids preceding the missing enzyme and those that do not require the missing enzyme (ie, build-up of compounds both before the block and "sideways" from the block). See following image. Treatment with exogenous glucocorticoid decreases ACTH secretion and subsequent suppression of overproduced steroids.
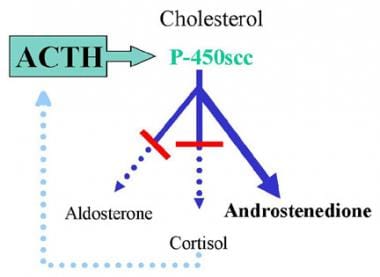 17-Hydroxylase Deficiency Syndrome. Representation of typical congenital adrenal hyperplasia (CAH). This example shows a deficiency in both the mineralocorticoid and glucocorticoid pathways. Decreased serum cortisol levels stimulate adrenocorticotropic hormone (ACTH) release via negative feedback. Increased ACTH secretion causes overproduction of adrenal steroids preceding the missing enzyme as well as those not requiring the missing enzyme. The example depicts a deficiency of 21-hydroxylase, resulting in deficient mineralocorticoid and glucocorticoid production and excessive androgen production.
17-Hydroxylase Deficiency Syndrome. Representation of typical congenital adrenal hyperplasia (CAH). This example shows a deficiency in both the mineralocorticoid and glucocorticoid pathways. Decreased serum cortisol levels stimulate adrenocorticotropic hormone (ACTH) release via negative feedback. Increased ACTH secretion causes overproduction of adrenal steroids preceding the missing enzyme as well as those not requiring the missing enzyme. The example depicts a deficiency of 21-hydroxylase, resulting in deficient mineralocorticoid and glucocorticoid production and excessive androgen production.
Cytochrome P450c17, an enzyme complex present in Leydig cells, ovarian follicles, and the adrenal zonae fasciculata and reticularis, catalyzes both 17-hydroxylase and 17,20 lyase activity. As might be expected from its location, P450c17 defects affect both adrenal and gonadal steroid production. P450c17 is the product of the cytochrome P45017 alpha gene (CYP17A1), and specific mutations of this gene cause varying degrees of partial-to-severe isolated 17-hydroxylase deficiency, isolated 17,20 lyase deficiency, or combined deficiencies. [2, 3, 4, 5, 6]
More than 100 different genetic mutations of the CYP17A1 gene have been described worldwide in patients with 17-hydroxylase deficiency [6, 7, 8, 9, 10, 11] For example, among the Chinese Han, two CYP17A1 mutations, D487-S488-F489 deletion and TAC329AA, account for the majority of 17-hydroxylase deficiency cases. [7, 12] Different CYP17A1 mutations have been found in other Chinese cases, including novel nonsense mutations R449C and L209P. [8] By contrast, in a Brazilian cohort of 19 families with 17-hydroxylase deficiency, [9] 7 different CYP17 mutations were found among 24 subjects. However, two mutations accounted for most cases: W406R (50%) and R362C (32%). In these families, phenotypic features varied among the subjects and did not correlate with the CYP17 genotype.
A rare cause of 17-hydroxylase deficiency syndrome, first reported in 2004, is autosomal recessive P450 oxidoreductase (POR) deficiency. POR is an obligate electron donor for all microsomal P450 enzymes, including P450c17 (17α-hydroxylase/17,20 lyase), P450c21 (21-hydroxylase) and P450 aro (aromatase). POR deficiency can affect multiple steroidogenic pathways and have variable presentations depending on relative degrees of impaired enzyme activity. Drug metabolism may also be affected in these patients as many drugs are metabolized by hepatic P450s. [13, 14, 15, 16]
C-17α-hydroxylase is necessary to convert pregnenolone to 17-hydroxypregnenolone (17-OH Preg) and progesterone to 17-hydroxyprogesterone (17-OH Prog) [1] ; see first image below. Thus, absence of this enzyme impairs all sex steroid and cortisol production (see second image below). Low levels of cortisol result in increased ACTH stimulation of steroids prior to the 17-hydroxylase step, resulting in increased accumulation and secretion of 17-deoxysteroids by the zona fasciculata, including pregnenolone, progesterone, deoxycorticosterone (DOC), and corticosterone (compound B).
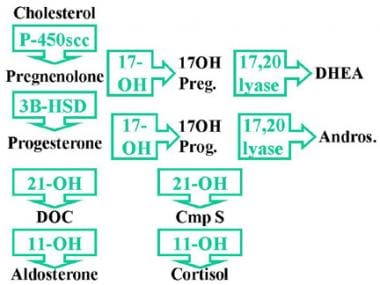 17-Hydroxylase Deficiency Syndrome. C-17α-hydroxylase is necessary to convert pregnenolone to 17-hydroxypregnenolone (17-OH Preg) and progesterone to 17-hydroxyprogesterone (17-OH Prog). Absence of C-17α-hydroxylase impairs all sex steroid and cortisol production. Low levels of cortisol result in increased adrenocorticotropic hormone (ACTH) stimulation of steroids prior to the 17-hydroxylase step, resulting in increased accumulation and secretion of 17-deoxysteroids by the zona fasciculata, including pregnenolone, progesterone, deoxycorticosterone (DOC), and corticosterone.
17-Hydroxylase Deficiency Syndrome. C-17α-hydroxylase is necessary to convert pregnenolone to 17-hydroxypregnenolone (17-OH Preg) and progesterone to 17-hydroxyprogesterone (17-OH Prog). Absence of C-17α-hydroxylase impairs all sex steroid and cortisol production. Low levels of cortisol result in increased adrenocorticotropic hormone (ACTH) stimulation of steroids prior to the 17-hydroxylase step, resulting in increased accumulation and secretion of 17-deoxysteroids by the zona fasciculata, including pregnenolone, progesterone, deoxycorticosterone (DOC), and corticosterone.
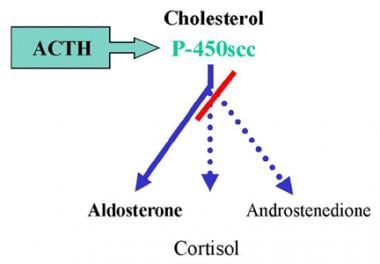 17-Hydroxylase Deficiency Syndrome. Graphic illustration of deficiency. Absence of C-17α-hydroxylase impairs all sex steroid and cortisol production.
17-Hydroxylase Deficiency Syndrome. Graphic illustration of deficiency. Absence of C-17α-hydroxylase impairs all sex steroid and cortisol production.
Hypogonadism occurs as a result of deficient sex steroid production. DOC mineralocorticoid activity causes sodium retention, plasma volume expansion, hypertension, hypokalemia, and decreased renin and aldosterone levels in most untreated patients with 17-hydroxylase deficiency.
Etiology
17-Hydroxylase (17-OH) deficiency syndrome is caused by mutations in the CYP17A1 gene located on chromosome 10q24.3. [17] More than 100 mutations have been identified [18] ; however, populations in which 17-OH deficiency syndrome is more prevalent, such as Brazilians, [9] Canadian Mennonites, [19] Dutch Frieslanders, [19] Japanese, Koreans, [10] and Chinese [20] have specific reoccurring mutations thought to be due to founder effect. [18]
A small number of CYP17A1 gene mutations have been found to cause isolated 17,20-lyase deficiency, which is characterized by abnormal sexual development without hypertension or hypokalemia. These mutations alter a region of the CYP17A1 protein that plays a role in the enzyme's 17,20-lyase function but not its 17α-hydroxylase function. As a result, 17,20-lyase activity is severely reduced but 17α-hydroxylase activity is normal. [21]
Epidemiology
Incidence
17-Hydroxylase deficiency is a rare cause of congenital adrenal hyperplasia (CAH), accounting for less than 1% of cases and occurring in 1 in 50,000 people globally. [22] Over 90% of individuals with CAH have 21-hydroxylase deficiency. [18] The incidence of classic 21-hydroxylase deficiency affects 1 in 16,000 live births. Milder (nonclassic) 21-hydroxylase deficiency is estimated to occur in 1 in 1,000 births and even more frequently in populations of specific ethnicities, such as Ashkenazi Jews (1 in 27), Hispanics (1 in 53), Yugoslavs (1 in 62) and Italians (1 in 300). [22, 23] The second most common type, 11-β -hydroxylase deficiency, represents 5-8% of CAH cases has an incidence of about 1 in 100,000 individuals but is 20 times higher in Moroccan Jews. [22] (See C-11 Hydroxylase Deficiency.)
17-Hydroxylase deficiency occurs worldwide. However, there have been fewer than 200 cases reported in the literature and most of these reported cases were either isolated or occurred in small clusters. Examples include Turkey, where the reported incidence was 1 in 273 patients with CAH over a 25-year period [24] ; Brazil, where 16 cases were reported over a 10-year period [9] ; and Puerto Rico, where 1 case was reported. [25] New cases of 17-hydroxylase deficiency continue to be reported, most recently in Korea and China. [10, 11, 20]
Age
A diagnosis of 17-hydroxylase deficiency may be suspected in infancy or childhood when hypokalemia and hypertension are found in association with either ambiguous genitalia or in an apparent female patient with a hernia or inguinal mass. [20] However, many patients may go undiagnosed until adolescence or young adulthood. Karyotypic 46,XY patients may be undiagnosed until puberty, having been raised as females, and present to an endocrinologist or nephrologist for evaluation due to lack of secondary sexual characteristics and varying degrees of hypertension and hypokalemia. Similarly, 46,XX patients are usually diagnosed upon presentation of delayed puberty or lack of menses, along with hypertension and hypokalemia. [26]
Prognosis
Prognosis for patients with 17-hydroxylase (17-OH) deficiency syndrome is usually good-to-excellent with adequate glucocorticoid therapy and monitoring. Patients rarely, if ever, have adrenal crises. Sex steroid replacement promotes development of secondary sexual characteristics in both sexes and cyclic menstrual bleeding in females. Fertility may be impaired in both male and female patients with severe deficiency. [27]
Corticosterone has some glucocorticoid activity; elevated levels (ie, 50-100 times normal) are adequate to prevent adrenal insufficiency. Thus, these patients do not have hypoglycemia, hypotension, or difficulties dealing with infections, stress, or surgical procedures. These patients also experience no virilization or accelerated prepubertal growth as is typical in more common types of congenital adrenal hyperplasia (CAH) that result from lack of sex steroids.
Most patients have some degree of hypokalemia and hypertension; blood pressure elevations range from mild to severe. Although 10-15% may have no hypertension or hypokalemia at presentation, patients may present with malignant hypertension or with severe, symptomatic hypokalemia. Hypertension sometimes may persist for months to years in older or more severely affected individuals, necessitating additional antihypertension therapy.
Complications
Despite sex steroid replacement, patients with 17α-hydroxylase/17,20-lyase deficiency develop hypergonadotropic hypogonadism and infertility due to the decreased enzymatic activity of CYP17A1.
Irreversible defects in steroidogenesis may lead to impaired spermatogenesis and folliculogenesis, and women are unable to conceive spontaneously or via endocrinological intervention. A single report of pregnancy in a patient with partial 17α-hydroxylase/17,20-lyase deficiency, resulted in the live birth of triplets following in vitro fertilization. [27] Two additional live births resulting from fertility therapy have been reported. [28, 29]
Although normal T production in males with 17OHD is possible, there are no reports of male fertility. Milder cases of 17OHD might be assumed to be “idiopathic hypospadias” if not evaluated thoroughly. Consequently, the capacity of men with partial 17OHD to reproduce might be higher than assumed from more severe cases. [27]
-
17-Hydroxylase Deficiency Syndrome. Normal adrenal steroid biosynthesis results in three products: mineralocorticoids (aldosterone), glucocorticoids (cortisol), and androgens (androstenedione). Cortisol production is regulated by feedback with adrenocorticotropic hormone (ACTH). ACTH stimulates the enzyme P-450scc (20,22 desmolase), with subsequently increased production of all adrenal steroids.
-
17-Hydroxylase Deficiency Syndrome. Representation of typical congenital adrenal hyperplasia (CAH). This example shows a deficiency in both the mineralocorticoid and glucocorticoid pathways. Decreased serum cortisol levels stimulate adrenocorticotropic hormone (ACTH) release via negative feedback. Increased ACTH secretion causes overproduction of adrenal steroids preceding the missing enzyme as well as those not requiring the missing enzyme. The example depicts a deficiency of 21-hydroxylase, resulting in deficient mineralocorticoid and glucocorticoid production and excessive androgen production.
-
17-Hydroxylase Deficiency Syndrome. C-17α-hydroxylase is necessary to convert pregnenolone to 17-hydroxypregnenolone (17-OH Preg) and progesterone to 17-hydroxyprogesterone (17-OH Prog). Absence of C-17α-hydroxylase impairs all sex steroid and cortisol production. Low levels of cortisol result in increased adrenocorticotropic hormone (ACTH) stimulation of steroids prior to the 17-hydroxylase step, resulting in increased accumulation and secretion of 17-deoxysteroids by the zona fasciculata, including pregnenolone, progesterone, deoxycorticosterone (DOC), and corticosterone.
-
17-Hydroxylase Deficiency Syndrome. Graphic illustration of deficiency. Absence of C-17α-hydroxylase impairs all sex steroid and cortisol production.






