Background
Holt-Oram syndrome (HOS) (OMIM 142900) is a heart–upper limb malformation complex with an autosomal dominant inheritance and near-complete penetrance but variable expression. Holt and Oram first described this syndrome in 1960. Approximately 40% of cases represent new mutations.
See the image below depicting Holt-Oram syndrome in an infant.
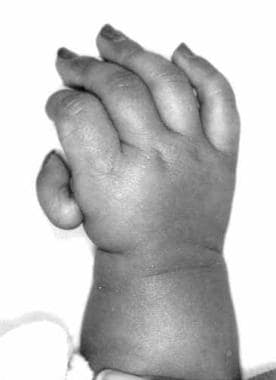 Photograph showing hypoplastic right thumb of the right hand of a 6-month-old infant with Holt-Oram syndrome.
Photograph showing hypoplastic right thumb of the right hand of a 6-month-old infant with Holt-Oram syndrome.
Pathophysiology
Defective development of the embryonic radial ray (eg, aplasia, hypoplasia, fusion, other anomalous development) results in a wide spectrum of phenotypes, including triphalangeal or absent thumbs, foreshortened arms, and phocomelia. The syndrome is associated with defective development of cardiac structures that results in atrial septal defect (ASD), most commonly the secundum type (as shown below); heart block of varying degree; or both. [1]
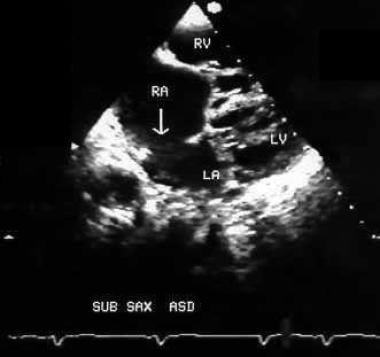 A 2-dimensional echocardiographic picture taken from subxiphoid window showing a large secundum atrial septal defect (arrow) in a 7-year-old boy with Holt-Oram syndrome. ASD = Atrial septal defect; RA = Right atrium; RV = Right ventricle; LA = Left atrium; LV = Left ventricle.
A 2-dimensional echocardiographic picture taken from subxiphoid window showing a large secundum atrial septal defect (arrow) in a 7-year-old boy with Holt-Oram syndrome. ASD = Atrial septal defect; RA = Right atrium; RV = Right ventricle; LA = Left atrium; LV = Left ventricle.
The responsible gene has been mapped to band 12q24.1, which encodes the human transcription factor TBX5. [2, 3, 4] A full list of the described mutations is available at the TBX5 Gene Mutation Database, an online locus-specific database that contains germline and somatic mutations of the TBX5 gene. One of the recently added loci is c.373G>A, which results in the missense mutation p.Gly125Arg; this is a novel mutation, in that it is associated with a gain-of-function mechanism and is associated with paroxysmal atrial fibrillation and no structural heart disease. [5, 6, 7]
TBX5 genotyping has high sensitivity and specificity for Holt-Oram syndrome (HOS) if stringent diagnostic criteria are used in assigning the clinical diagnosis. Mutations of this gene introduce a premature stop codon and result in truncated protein versions. Consequent abnormal expression of the cardiac and limb-specific T-box transcription factors lead to the malformations described in HOS. The T-box gene family is a group of related genes that play a critical role in human embryonic development. [8, 9]
A cardiomelic developmental field has also been postulated to relate the genetic heterogeneity of HOS (and other similar syndromes) to a cascade of molecules, including the brachyury, sonic hedgehog, bone morphogenetic protein, retinoic acid receptor, and transforming growth factor beta families.
Disturbed fetal limb muscle development has also been reported and may underlie the bony malformations.
Expression widely varies in different generations. Ogur et al reported variable clinical expression of HOS in three generations. [10] The grandfather presented with phocomelia of arms, with three digits on each hand, congenital heart defect, and narrow shoulders. His son presented with cardiac conduction disturbance with no congenital heart or skeletal defect. His granddaughter developed ventricular septal defect (VSD) and moderate radial deviations of both hands, with no obvious hypoplasia of the extremities. Cachat et al reported a father and two sons in a French family with HOS who presented with different types of ASDs: ostium primum ASD, secundum ASD, and sinus venosus ASD, respectively. [11]
Malignant hyperthermialike manifestations have been reported in a 2-month-old child with HOS undergoing cardiac surgery; however, the association or mechanism has yet to be identified. [12]
Epidemiology
United States data
The incidence rate of Holt Oram syndrome (HOS) is unknown.
International data
In a 2014 report, the mean prevalence of HOS diagnosed prenatally or in the early years of life in European registries was 0.7 per 100,000 births or 1:135,615 births. [13]
In Hungary, the birth prevalence is 0.95 per 100,000 total births. About 350 cases have been reported worldwide. A report identified this syndrome in 4% of patients with radial longitudinal deficiency. [14]
Race-, sex-, and age-related demographics
No valid racial data are available.
Both sexes are equally affected, although the defects tend to be more severe in females.
HOS malformations are present at birth. Age at presentation varies according to the extent of the abnormality externally visible and the type of associated heart defect, if any.
Prognosis
Prognosis in patients with Holt-Oram syndrome (HOS) is dictated by the severity and type of cardiac and limb malformations. Because the most common defect in HOS is atrial septal defect (ASD), the prognosis is excellent. [15]
A scoring system to assess severity has been recommended by Gall et al and modified by Gladstone and Sybert (see Presentation).
Mortality/Morbidity
No valid figures are available because the condition, in and of itself, has no specific mortality or morbidity. The mortality and morbidity relate directly to the associated congenital abnormalities, particularly those of the heart. For example, mortality and morbidity of a secundum atrial septal defect (ASD) is negligible throughout childhood, including patients who undergo procedures to close the ASD. Please see the appropriate respective articles for mortality and morbidity figures on specific cardiac defects.
Causes of death include cardiac malformation and heart block. See the respective article for each cardiac abnormality for a discussion of cause of death.
Complications
Complications secondary to heart disease and heart failure include tachyarrhythmia, especially atrial fibrillation [5, 16] and conduction abnormalities place patients at special risk during anesthetic procedures.
Complications secondary to limb malformation include the following:
-
Those secondary to interventions that may be required
-
Contractures and deformity
Psychological problems may arise secondary to disability.
Patient Education
Parents and patients
Parents and patients should understand the various manifestations and should undergo genetic counseling. Note the following:
-
In any child with atrial septal defect (ASD), the patient and parents should be carefully examined for limb malformations, and a family history should be studied in detail.
-
Detection of subtle limb defect alters the recurrence risk in offspring from the empirical risk of an isolated ASD (3%) to that for an autosomal dominant trait (50%).
Patient should avoid any activity beyond tolerance as well as avoid postures that might lead to deformities.
Prenatal counseling
Note the following:
-
Prospective parents should be alerted to the fact that a child born with Holt-Oram syndrome to an affected parent has a 1 in 3 chance of having a severe reduction abnormality of the upper limb, with a 1 in 22 risk of phocomelia.
-
If an ASD is present, the risk of serious limb abnormality is greater than if a VSD or conduction defect occurs alone.
-
Severity is likely to be greater if the transmitting parent is female.
-
The detection of a severe reduction defect before or after birth indicates a high probability of an associated structural cardiac lesion.
-
Photograph showing hypoplastic right thumb of the right hand of a 6-month-old infant with Holt-Oram syndrome.
-
Photograph of the left hand of a 6-month-old infant with Holt-Oram syndrome showing total aplasia of the left thumb.
-
Plain radiograph of the right forearm and hand of a 5-month-old infant with Holt-Oram syndrome showing hypoplastic radius and ulna and only 4 metacarpals.
-
A 2-dimensional echocardiographic picture taken from subxiphoid window showing a large secundum atrial septal defect (arrow) in a 7-year-old boy with Holt-Oram syndrome. ASD = Atrial septal defect; RA = Right atrium; RV = Right ventricle; LA = Left atrium; LV = Left ventricle.
-
Color Doppler echocardiographic picture taken from subxiphoid window showing the large left-to-right flow of blood (arrow) across the atrial septal defect. The red color pattern depicts flow direction from left atrium (LA) to right atrium (RA). ASD = Atrial septal defect; RA = Right atrium; RV = Right ventricle; LA = Left atrium; LV = Left ventricle.





