Background
Hirschsprung disease, a common cause of neonatal and infantile large gut obstruction, [1] was first described in 1886 by Harold Hirschsprung as a cause of constipation in early infancy. This disease is characterized by a variable extent of contiguous aganglionosis extending from the anorectum proximally. [1] Early recognition and surgical correction of Hirschsprung disease protects affected infants from enterocolitis and debilitating constipation.
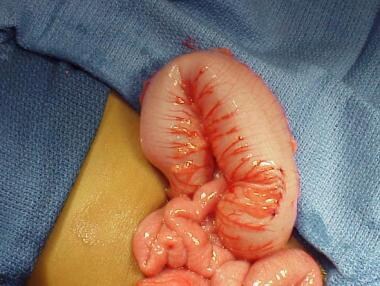 Pediatric Hirschsprung disease. Intraoperative finding in total colonic aganglionosis. Note the decompressed bowel adjacent to the distended colon.
Pediatric Hirschsprung disease. Intraoperative finding in total colonic aganglionosis. Note the decompressed bowel adjacent to the distended colon.
Pathophysiology
Hirschsprung disease results from the absence of enteric neurons within the myenteric and submucosal plexus of the rectum and/or colon. [2] Enteric neurons are derived from the neural crest and migrate caudally with the vagal nerve fibers along the intestine. These ganglion cells arrive in the proximal colon by 8 weeks' gestation and in the rectum by 12 weeks' gestation. Arrest in migration leads to an aganglionic segment. This results in clinical Hirschsprung disease.
Etiology
Genetic causes
The disease is generally sporadic, although the incidence of familial disease has been increasing (overall incidence: 7.6% [3] ). Hirschsprung disease is also reported to occur in two thirds of twins, primarily males. [3]
Multiple loci appear to be involved, including chromosomes 13q22, 21q22, and 10q.
Mutations in the Ret proto-oncogene have been associated with multiple endocrine neoplasia (MEN) 2A or MEN 2B and familial Hirschsprung disease. [4, 5]
Other genes associated with Hirschsprung disease include the glial cell-derived neurotrophic factor gene, the endothelin-B receptor gene, and the endothelin-3 gene.
Associated conditions
Hirschsprung disease is strongly associated with Down syndrome; 5-15% of patients with Hirschsprung disease also have trisomy 21.
Other associations include Waardenburg syndrome, congenital deafness, malrotation, gastric diverticulum, and intestinal atresia.
Epidemiology
United States and international data
Hirschsprung disease occurs in approximately 1 per 5000 live births. [6]
The worldwide prevalence may vary by region and has been shown to be as high as 1 per 3000 live births in the Federated States of Micronesia. [7]
Sex- and age-related demographics
Hirschsprung disease is approximately four times more common in males than females. [8]
Nearly all children with Hirschsprung disease are diagnosed during the first 2 years of life. Approximately one half of children affected with this disease are diagnosed before they are aged 1 year. A small number of children with Hirschsprung disease are not recognized until much later in childhood or adulthood.
Prognosis
The outcome in infants and children with Hirschsprung disease is generally quite good. Most children obtain fecal continence and control. However, children with other significant comorbidities, such as major genetic abnormalities, may have lower rates of continence. [9]
Morbidity/mortality
Left untreated, Hirschsprung disease is fatal due to bacteremia and then sepsis in the context of bowel inflammation (enterocolitis) or bowel perforation. [2]
The overall mortality of Hirschsprung enterocolitis is 25-30%, which accounts for almost all of the mortality from Hirschsprung disease.
Total colonic aganglionosis occurs in an estimated 2-13% of patients with Hirschsprung disease, and may extend proximally from the colon into varying lengths of the small bowel. [10]
Patient Education
Alert patients and their families to potential preoperative and postoperative complications of Hirschsprung disease. When applicable, teach patients and their families how to care for an ostomy.
-
Pediatric Hirschsprung disease. Abdominal radiograph demonstrating small bowel obstruction and megacolon in an infant with Hirschsprung disease.
-
Pediatric Hirschsprung disease. Barium enema demonstrating the transition zone. The transition zone shows the transition from dilated, normally innervated bowel to normal caliber, noninnervated bowel.
-
Pediatric Hirschsprung disease. Intraoperative finding in total colonic aganglionosis. Note the decompressed bowel adjacent to the distended colon.









