Background
Angioedema is a subcutaneous extension of urticaria, resulting in deep swelling within subcutaneous sites. In contrast, urticaria results from transient extravasation of plasma into the dermis, causing a wheal characterized by tense edema with or without redness.
Angioedema can occur with generalized urticaria if the tissue swelling has indistinct borders around the eyelids and lips. In addition, when the swelling of urticaria extends to the face, hands, feet, and genitalia, the clinical manifestation may be called angioedema. As many as 50% of children who have urticaria exhibit angioedema, with swelling of the hands and feet.
Classification of angioedema
Based on the current clinical understanding of angioedema, dividing cases into the following types is useful:
-
Hereditary angioedema type 1 (HAE1)
-
Hereditary angioedema type 2 (HAE2)
-
Hereditary angioedema type 3 (HAE3)
-
Acquired angioedema type 1 (AAE1) (very rare; only a few reported cases)
-
Acquired angioedema type 2 (AAE2) (very rare; only a few reported cases)
-
Nonhistaminergic angioedema (INAE) (may occur in approximately 1 of 20 angioedema cases)
-
Idiopathic angioedema
-
Allergic angioedema (most common form)
-
ACE inhibitor–induced angioedema (4-8% of cases)
Hereditary angioedema (HAE) accounts for only 0.4% of angioedema cases; however, the specific diagnostic tests and high mortality rate associated with hereditary angioedema deserve special attention.
In 1876, Milton described the first case of hereditary angioedema. Six years later, Quincke introduced the term angioneurotic edema to describe this disease. Later, Osler described the disease as episodic bouts of well-circumscribed nonpitting subepithelial edema that primarily involved the extremities, larynx, face, and abdomen.
Hereditary angioedema is an autosomal dominant disease usually associated with a positive family history of angioedema. However, numerous cases are due to a new mutation of the gene.
In approximately 80-85% of hereditary angioedema cases, serum levels of C1 inhibitor (C1INH) are decreased to approximately 30% of reference range values. In contrast, about 15% of patients with hereditary angioedema have reference range levels of antigenic, but mostly nonfunctional, C1INH. Missing or nonfunctional C1INH leads to failure in controlling the enzymatic activity of C1, resulting in lower levels of the early-acting complement components C4 and C2 because of overconsumption.
No close relationship between plasma C1INH levels and the severity of attacks has been noted. Some patients with very low C1INH levels have few attacks, whereas others with much higher levels of C1INH have much more severe disease.
HAE1 (low levels of functional C1INH) may be due to a wide range of gene mutations. In HAE2 (reference range or even increased levels of antigenic but nonfunctional C1INH), different point mutations have been described within or near the reactive center of the C1INH gene (SERPING1). (See the image below.)
The structural abnormalities in the SERPING1 genes in patients with hereditary angioedema have been found to be very heterogeneous. More than 150 mutations have been reported in unrelated patients. Agostino et al have reported on the details of genetic analysis. [1]
HAE3 is the most recently described type of hereditary angioedema. In HAE3, C1INH function and complement components are normal. [2] Mutations in the gene that encodes coagulation factor XII (Hageman factor) have been found in some patients with HAE3.
Essential features of HAE3 include the following:
-
A long history of recurrent skin swelling, attacks of abdominal pain, or episodes of upper airway obstruction
-
Familial occurrence, exclusively in female members of the family
-
No history of urticaria in the patient or other family members
-
Normal C1INH and C4 concentrations in the plasma
-
Failure to respond to antihistamines, corticosteroids, or C1INH concentrate
AAE1 is usually linked to an underlying lymphoproliferative disorder. Complement-activating factors, idiotype/anti-idiotype antibodies, or other immune complexes associated with the underlying disorder destroys the function of C1INH. The onset of angioedema can precede other symptoms of a lymphoproliferative disease; thus, exploring the possibility of underlying malignancy in cases of AAE1 is vital.
AAE2 is associated with autoantibodies that directly inhibit C1INH function. No underlying disorder is apparent. AAE1 and AAE2 are very rare in the pediatric population.
INAE angioedema is angioedema without urticaria. Patients usually do not respond to H1 blockers (antihistamines). Parasites, infections, and autoimmune diseases are not present.
The idiopathic form of angioedema may be associated with swelling, hives that persist longer than 6 weeks, or both. Thyroid dysfunction should be considered.
Allergic angioedema is characterized by swelling, hives, or both in reaction to environmental factors such as food, an insect sting or bite, cold, heat, latex, or a drug. Usually, these environmental factors provoke histamine release that leads to swelling, hives, or both.
Angioedema can also be caused by ACE inhibitors (eg, captopril, enalapril, genzapril, quinapril, ramipril) used to treat high blood pressure. Swelling may begin a few hours to years after first starting the medication.
For other discussions on angioedema, see the overview topics Acquired Angioedema and Hereditary Angioedema.
Pathophysiology
The pathophysiology of urticaria-associated angioedema is fully discussed in Urticaria.
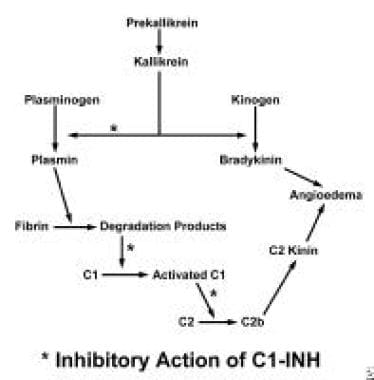
The cause of angioedema in patients with hereditary angioedema is still unknown. One hypothesis involves persistent activation of C1, resulting in ongoing cleavage of the next 2 components of the complement cascade, C4 and C2. According to this hypothesis, cleaved C2 is acted on by other proteolytic enzymes (possibly plasmin), generating a kinin-like molecule that causes angioedema. Involvement of local mediators is virtually uncertain.
A second hypothesis is that angioedema attacks are caused by activation of the kinin-generating system, which involves cleavage of high-molecular-weight kininogen by activated kallikrein with attendant formation of bradykinin. Bradykinin is believed to be responsible for angioedema episodes.
In hereditary angioedema, 2 phenotypic variants have been described. HAE1 may be due to a wide range of gene mutations, resulting in either a lack of messenger RNA transcription or transcription in abnormal messenger RNAs that are not translated into a stable protein. In HAE2, different point mutations have been described within or near the reactive center, resulting in different dysfunctional proteins.
AAE is due to the production of a C1INH-consuming factor by malignant cells. In fact, most cases of acquired C1INH-deficient angioedema have been associated with the presence of a lymphoid or other malignancy. In rare cases, C1INH deficiency could be due to consumption by the immune complex during the course of an autoimmune disease. Another type of C1INH deficiency is the result of monoclonal or oligoclonal production of antibodies that appear to recognize C1INH and destroy its functional activity.
The fluctuations in sex hormone levels at the beginning of adolescence, in the perimenopausal period, during pregnancy, or during the use of oral contraceptives can precipitate edematous attacks in hereditary angioedema. One study indicated that the number of attacks was significantly higher in females with high progesterone levels (≥4 nmol/L); a significantly lower attack frequency was noted in patients with a higher (40 nmol/L) sex hormone–binding globulin (SHBG) level. [3] Thus, monitoring these 2 hormonal levels may be useful in predicting attacks in patients with hereditary angioedema.
Studies on the function of C1INH have helped further the understanding of angioedema pathophysiology. C1INH controls activation of the complement system by inhibiting the esterase activity of C1r and C1s in the classic pathway and the esterase activity of mannan-binding lectin serine peptidase 2 (MASP2) in the mannose-binding lectin pathway.
The second major physiological role of C1INH is now believed to be regulation of the contact system, where it intervenes and inhibits activated coagulation factor XII and kallikrein. In addition to these 2 major functions, C1INH inhibits factor XI, plasmin, and the tissue plasminogen activator (tPA). The relevance of these activities in vivo remains controversial. However, convincing in vivo evidence supports the activation of plasminogen in humans by factor XIIa.
HAE3 has been exclusively observed in women, in whom it appears to be correlated with high estrogen levels (eg, pregnancy, the use of oral contraceptives). One report proposed 2 missense mutations (ie, c.1032C→A and c.1032C→G) in F12, the gene that encodes human coagulation factor XII (Hageman factor), as a possible cause of HAE3. [4] Transcription of F12 is positively regulated by estrogens, which may explain why only women are affected by HAE3.
Another extensive family study of HAE3 was reported. In this family a missense mutation Thr309Lys was identified in the factor XII gene with a heterozygotic pattern. [5] This mutation was also identified in the mother of the patient, her daughter, and her son.
Along with the 5 German and French families that have been reported for HAE3, a large Italian family was also reported from Canada. [6] A missense mutation in F12 was present in the 3 affected female subjects of this family who had estrogen-dependent inherited angioedema.
In addition, these affected females have polymorphisms associated with lower levels of both aminopeptidase and angiotensin-1 converting enzyme, the major enzymes responsible for bradykinin degradation. Thus, multiple genes may contribute to estrogen-dependent or estrogen-associated angioedema, leading to the observed heterogeneity of clinical features.
Etiology
Hereditary angioedema (HAE) is an autosomal dominant condition. In approximately 50% of cases, clinical manifestations may appear during childhood. [7] Risk factors for HAE episodes include trauma, such as surgery, dental manipulation, or accidents. Episodes of HAE are the result of unopposed complement activation and/or activation of the kinin generating system due to the C1INH deficiency.
In HAE type 3, a fluctuation of sex hormones has been speculated to precipitate HAE attacks.
In 2010, the international consensus algorithm for the diagnosis, therapy, and management of HAE was updated. [8] The consensus approach is only an interim guide to HAE, which is a complex disorder. This consensus should be replaced as soon as possible with large phase lll and IV clinical trials, meta-analyses, and database registry validation of approaches, including quality-of-life and cost-benefit analyses, followed by large head-to-head clinical trials and then evidence-based guidelines and standards for HAE disease management.
Diagnosis and treatment of HAE with normal C1 inhibitor (originally reported as HAE type 3) was recently reviewed by Bork. [9] Most of the patients were women. In many of the affected women, oral contraceptives, hormone therapy containing estrogens, and pregnancies triggered the clinical symptoms. Recently, in some families, mutations in the coagulation factor Xll (Hageman factor) gene were detected in the affected persons.
Acquired forms of angioedema are commonly associated with lymphoproliferative disorders or malignancy. They are relatively uncommon in pediatric patients. The known cause of angioedema has been increasingly reported in clinical practice. Therefore, it is prudent to consider those possibilities when inciting agents are used on patients.
Patients taking ACE inhibitors, which are rarely used in children, may experience episodic angioedema. The cause of acquired angioedema in patients using ACE inhibitors is believed to be the accumulation of bradykinin, which is subject to breakdown by ACE. ACE inhibitors help build up bradykinin levels in the tissue.
A severe case of angioedema was reported in a patient who received treatment with rituximab for rheumatoid arthritis. [10] Angioedema was reported after intravitreal bevacizumab was injected into the eye for the treatment of cystoid macular edema. The angioedema recurred when the patient received the same drug in the other eye later. It was the first report of a systemic reaction with this agent. [11]
Angioedema was reported in a female when she had the first exposure to hair dye. It contained paraphenylenediamine (PPD). Related compounds have been used in black henna tattoos, are widely used. [12]
Bedbug (insect family Cimicidae) infestation is now known to cause systemic reactions, including angioedema. Thus, an integrated pest management strategy should be used to eliminate infestation. [13]
Epidemiology
Urticaria-associated angioedema occurs in nearly 50% of children who have urticaria. Because urticaria occurs in 2-3% of children, urticaria-associated angioedema is estimated to occur in 1-2% of the general population.
The frequency rate of hereditary angioedema diseases is currently unknown. Current estimates suggest that the disease affects 1 in 10,000-50,000 persons. An estimated 5,000 to 25,000 patients in the United States have HAE1. The exact frequency of HAE2 is unknown, but the ratio of HAE1 to HAE2 is 85:15. In the past 5 years, the number of spontaneous mutations in newly diagnosed hereditary angioedema cases has increased 50%, indicating the need for more accurate epidemiologic data.
No epidemiologic data are available for acquired C1INH deficiency because these patients have only been described in case reports. No epidemiologic data are currently available regarding patients with HAE3.
Race-, sex-, and age-related demographics
No racial differences are known in most forms of angioedema. However, African Americans are at increased risk for ACE inhibitor–related angioedema.
Urticaria-related angioedema has no known sex differences. Hereditary angioedema is an autosomal dominant disorder and affects both sexes. HAE3 has been exclusively reported in females; the influence of an X-linked gene on the generation of vasoactive peptides has been speculated.
Urticaria-related angioedema has no known age differences. Clinical manifestations of hereditary angioedema are more common beyond the teenaged years.
Prognosis
Urticaria-associated angioedema is generally self-limited in pediatric patients. In patients with hereditary angioedema, the onset of symptoms frequently occurs during the teenage years. Morbidity varies from case to case. In some patients, acute attacks occur once every several years, whereas attacks in others occur several times a year. Morbidity changes after therapy begins.
Sudden onset of severe laryngeal edema can lead to death. In the past, the mortality rate for attacks involving the upper airways exceeded 25%. Severe abdominal pain may sometimes subject the patient to unnecessary surgery.
Patient Education
The following aspects of patient education should be considered:
-
Patients should be closely monitored by specialists for lifelong care
-
Patients should be careful to avoid trauma
-
Patients should wear a MedicAlert bracelet or carry an identification card at all times
-
Patients should keep up with prophylactic medication
More information is available from the Hereditary Angioedema Association.
For patient education information, see the Allergy Center and Skin, Hair, and Nails Center, as well as Hives and Angioedema.
-
The mechanism of angioedema resulting from C1-esterase inhibitor deficiency.
-
Angioedema secondary to ACE inhibitors
Tables
| Generic (Brand) | Population | Route |
Mechanism |
|---|---|---|---|
C1 inhibitor, plasma derived (Cinryze) |
Aged 6 y and older |
IV | Inhibits plasma kallikrein, coagulation factors Xlla, XIIf, and Xla, C1s, C1r, MASP-1, MASP-2, and plasmin |
C1 inhibitor, plasma derived (Haegarda) |
Aged 6 y and older |
SC | Inhibits plasma kallikrein, coagulation factors Xlla, XIIf, and Xla, C1s, C1r, MASP-1, MASP-2, and plasmin |
Lanadelumab (Takhzyro) |
Aged 2 y and older |
SC | Kallikrein inhibitor |
Berotralstat (Orladeyo) |
Adolescents and adults |
PO | Kallikrein inhibitor |
| Danazol | Adults and pediatrics (off-label) |
PO | Androgen; mechanism unknown |
| Oxandrolone | Off-label | PO | Androgen; mechanism unknown |
| Methyltestosterone | Off-label | PO | Androgen; mechanism unknown |
Aminocaproic acid (Amicar) |
Off-label | PO | Inhibits plasminogen activation and plasmin activity |
Tranexamic acid (Cyklokapron) |
Off-label | PO | Inhibits plasminogen activation and plasmin activity |
| Generic (Brand) | Population | Route | Mechanism |
|---|---|---|---|
Ecallantide |
Adolescents and adults |
SC | Kallikrein inhibitor |
Icatibant |
Adults | SC | Bradykinin B2 antagonist |
C1 inhibitor, recombinant |
Adolescents and adults |
IV | Inhibits plasma kallikrein, coagulation factors Xlla, XIIf, and Xla, C1s, C1r, MASP-1, MASP-2, and plasmin |
C1 inhibitor, plasma derived |
Children and adults |
IV | Inhibits plasma kallikrein, coagulation factors Xlla, XIIf, and Xla, C1s, C1r, MASP-1, MASP-2, and plasmin |