Background
Pierre Robin sequence (PRS; also referred to as Pierre Robin malformation, Pierre Robin malformation sequence, Robin sequence, Pierre Robin syndrome, and Pierre Robin anomalad) is a condition consisting of three essential components [1, 2] :
-
Micrognathia or retrognathia
-
Cleft palate (usually U-shaped but sometimes V-shaped)
-
Glossoptosis, often accompanied by airway obstruction
The condition is named after the French dental surgeon Pierre Robin (1867-1950). His first paper described only micrognathia, glossoptosis, and respiratory distress; a decade later, he included cleft palate in the list of symptoms. His main interest was glossoptosis; over a 30-year period, he published more than 20 articles and monographs on the embryology, anatomy, complications, and management of this disorder.
PRS is a series of anomalies all initiated by one developmental problem. Other definitions have been suggested, based on a combination of mandibular deficiency, presence of U-shaped or V-shaped cleft palate, and airway obstruction.
PRS is not only causally heterogenous but also pathogenetically and phenotypically variable. [3] Each of the three symptoms can occur along a wide spectrum of severity. On the basis of the combination of symptoms, newborn babies and infants suffer from airway obstruction and feeding problems. An approach dividing this condition into three grades of severity has been recommended. [4]
PRS occurs as an isolated defect, as part of a recognized syndrome, or as part of a complex of multiple congenital anomalies. Diagnosis of a possible syndrome is very often critically important for correct management of a newborn affected with PRS. [5, 6]
Pathophysiology
PRS is etiologically heterogenous. This etiologic heterogeneity suggests pathogenetic heterogeneity and phenotypic variability, which include various causes of malformations and deformations and connective tissue dysplasia (see Etiology). A major distinction should be made between isolated occurrences of PRS and cases in which PRS is part of a recognized syndrome, part of a complex of multiple anomalies, or part of an unrecognized syndrome.
Isolated PRS is often a deformation resulting from intrauterine forces acting on the mandible, which restrict its growth and impact the tongue between the palatal shelves. Some deformational cases of PRS have been associated with oligohydramnios. Because micrognathia results from intrauterine molding, mandibular catchup growth is expected after birth once intrauterine forces are removed. The most severe cases of micrognathia are unlikely to be isolated PRS caused by deformation. Therefore, catchup growth is unlikely.
In patients with PRS, 13-27.7% of other family members are affected by cleft lip with or without cleft palate. [7, 8] Jakobsen et al compared data in several databases and proposed genes that might participate in the etiology of PRS, including GAD67 on 2q31, PVRL1 on 11q23-q24, and SOX9 on 17q24.3-q25.1. [9] Melkoniemi et al detected disease-associated mutations in COL11A1 and COL11A2 genes in some patients with nonsyndromic PRS. [10]
Jakobsen et al screened 10 unrelated patients affected with PRS for SOX9 and KCNJ2 mutations and suggested that nonsyndromic PRS may be caused by both SOX9 and KCNJ2 dysregulation. [11] Several lines of evidence for the existence of a 17q24 locus underlying PRS, including linkage analysis results, a clustering of translocation breakpoints 1.06-1.23 Mb upstream of SOX9, and microdeletions both approximately 1.5 Mb centromeric and approximately 1.5 Mb telomeric of SOX9, were reported by Benko et al. [12]
The proportion of cases that are isolated PRS varies in different studies. Hanson et al found that 25% of PRS cases had specific syndromes, another 35% had multiple anomalies without a specific recognized syndrome, and only 40% had isolated PRS. [13] Another study found that 74% of cases were isolated PRS. [14]
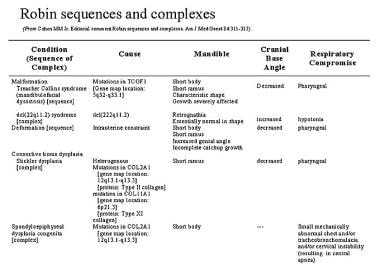
Among syndromic varieties of PRS, the most common is Stickler syndrome, which accounts for 20-25% of all cases. The second most common PRS syndrome is velocardiofacial syndrome, which accounts for about 15% of all PRS cases. [15] Treacher Collins syndrome (mandibulofacial dysostosis), Nager syndrome, spondyloepiphyseal dysplasia congenita (SEC), and other recognized syndromes account for the rest of the syndromic PRS cases.
Cohen listed 46 conditions associated with PRS (see the image below). [16] Although this list is representative, it is not complete. PRS may be present with other conditions and various other anomalies, especially those involving the eye, ear, heart, and limb. [17]

When Robin sequence is diagnosed, a full genetic evaluation (including fluorescence in situ hybridization [FISH] for 22q deletion and testing for mutation in the Treacle [TCOF1] gene) is appropriate, together with diagnostic tests for other suspected syndromes (eg, bone radiographs and ophthalmologic examination).
Among 47 patients with PRS who were monitored by Sheffield et al, 12 patients were diagnosed as syndromic. [18] Out of 52 cases reported by Sher, 15 patients had Stickler syndrome and only five had nonsyndromic PRS. [19]
Distinguishing between micrognathia (ie, a small mandible) and retrognathia (ie, an essentially normal-sized mandible in an abnormal position) is important. In micrognathia, the mandible is small; in retrognathia, the mandible is essentially normal in size but is retrognathic in position because the cranial base angle is larger than normal. Most PRS conditions are either micrognathic or retrognathic. [20]
In velocardiofacial syndrome, the mandible is retrognathic. Because the cranial base is altered, the mandible grows downward instead of forward. This gives the appearance of a small mandible, but the bone mass is normal. Retrognathia rarely produces severe airway distress.
In the vast majority of other syndromes, the mandible is micrognathic. The bone mass is decreased, and the mandible is disproportionately small. Severe airway obstruction is more common with these syndromes.
One of the most severe problems with airway obstruction may occur in patients affected with SEC. Cleft palate or PRS is often present in this autosomal dominant condition that has a mutation in the COL2A1 gene, located on chromosome 12 (12q13.11-q13.2), the same as that found in Stickler syndrome type I (hereditary progressive arthro-ophthalmopathy).
The respiratory compromise in SEC is caused by multiple mechanisms, including a small abnormal chest, a tracheobronchomalacia, and a central apnea caused by compression of cervical spinal cord or medulla oblongata from cervical instability. [21] Furthermore, the upper respiratory obstruction of PRS may worsen the respiratory condition of the patient with SEC (see the image below).
The mandible in PRS is often compared with the mandible in Treacher Collins syndrome (see the image below). When comparing two newborn babies with these two conditions, one can see that the mandible is short in both.
Because the severity of the defects varies widely in both conditions, the defects in PRS may initially seem much greater than those in Treacher Collins syndrome; however, a significant difference between these conditions becomes very apparent as the infant develops. In deformational PRS, so-called catchup growth occurs, though it may be incomplete. [22] In Treacher Collins syndrome, mandibular growth remains severely affected.
Etiology
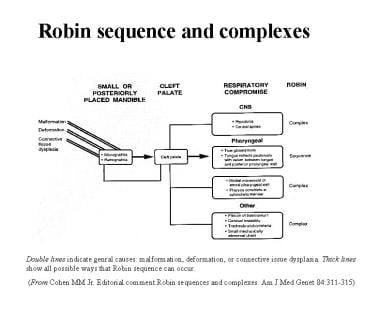
Suggested causes of PRS and Robin complexes include malformation, deformation, and connective-tissue dysplasia. [23] Because of differences in pathogenetic causes and phenotypes, various forms of PRS or Robin complexes can occur (see the image below).
For example, a pure exogenous factor such as oligohydramnios causing mandibular constraint leads to a failure of the tongue to descend and starts a sequence that ends as PRS. However, intrinsic intrauterine mandibular hypoplasia that may be part of a complex of anomalies (syndrome) caused by a chromosomal aberration can cause the same problem (ie, failure of tongue descent) and end in exactly the same way as the previous example; yet the former is a deformation sequence and the latter is a malformation sequence.
Epidemiology
The reported birth prevalence of PRS has ranged from 1 in 2000 to 1 in 30,000. [24] Bush and Williams suggested 1 in 8500. [25] The case definition of PRS still varies, and differences in definition lead to differences in the reported birth prevalence. [26] In studies with the highest birth prevalence of PRS, syndromic cases are most likely included.
A study of a population-based sample of 4433 patients with orofacial cleft (ascertained from 2,509,881 California births) reported the birth prevalence of nonsyndromic PRS to be 1 in 18,730 (~0.05 cases per 1000 births). [27]
Most nonsyndromic PRS cases are sporadic. In the older literature, several authors reported a familial occurrence [28, 29, 30] ; some of these cases were probably syndromic. The authors' recommend that Stickler syndrome be considered first when a familial occurrence of PRS is found. Stickler syndrome is the most common syndrome among PRS cases, and PRS is the most constant feature of Stickler syndrome. When correctly diagnosed, myopia is detected early; this can prevent retinal detachment and possibly blindness.
Prognosis
All neonates with significant PRS are at risk for sudden death. The sudden infant death syndrome (SIDS) data show that the risk of SIDS is increased when infants sleep in the prone position. Neonates with PRS already have a compromised airway, and they also typically require prone positioning. Accordingly, monitoring of these neonates should be strongly considered.
Infants with PRS deserve to be treated with a multidisciplinary approach that involves a knowledgeable and experienced team capable of providing a comprehensive assessment, a realistic plan of treatment, and appropriate follow-up. Engaging the family in the early stages of the evaluation, the ongoing medical investigations, the issues regarding the child's care, and future planning generally leads to satisfaction, even in the most difficult of medical scenarios.
In a study of 103 patients followed for a median of 8.6 years (range, 0.1-21.9 y), Logjes et al documented a 10% mortality (n = 10) at a median patient age of 0.8 years (range, 0.1-5.9 y). [31] Of the 10 infants who died, nine had syndromic PRS; seven of the nine died of respiratory insufficiency due to various causes, and the other two died of arrhythmia due to hypernatremia and of West syndrome with status epilepticus. The infant with nonsyndromic PRS died of brain ischemia after mandibular distraction osteogenesis.
In a study that included 67 neonates with PRS, Khansa et al compared the airway and feeding outcomes of three treatment approaches: mandibular distraction osteogenesis (n = 29), tongue-lip adhesion (n = 19), and conservative management without surgery (n = 19). [32] At follow-up, the mean apnea-hypopnea index (AHI) was similar across the three groups, as was the percentage of patients in each group who achieved an AHI of 5 or lower. At 1 year, the three groups showed no significant differences in weight percentiles or in risk of failure to thrive. One patient from the tongue-lip adhesion group required a tracheostomy.
Patient Education
Children with PRS have difficulties with feeding. [33] A cleft palate prevents production of the negative pressure necessary for sucking during breastfeeding. In addition, because of an abnormal jaw position, a baby with a small mandible usually has difficulties contracting its orbicularis oris muscle and squeezing the mother's nipple. In cleft palate, a wide communication between the oral and nasal cavities creates a risk of aspiration, nasal regurgitation, choking, and other feeding problems.
Consultation with a feeding specialist is advised. In many cases, when carefully instructed, a mother is able to manage bottle feeding while her baby is in a semisitting position. Special cleft palate nipples and squeezing bottles are helpful (see Cleft Lip and Palate). In patients with severe problems, gavage feeding may be necessary in the beginning of the baby's life.
Verifying that the mother of the baby with PRS is familiar with emergency techniques for the prevention of suffocation by food, such as the Heimlich maneuver, is important.
Genetic counseling may be helpful for parents. [34]
-
Three-week-old baby boy affected with nonsyndromic Pierre Robin sequence.
-
One-month-old baby affected with nonsyndromic Pierre Robin sequence.
-
Child with nonsyndromic Pierre Robin sequence at age 4 years. Profile is almost normal because of catchup growth.
-
Child with nonsyndromic Pierre Robin sequence at age 4 years.
-
U-shaped and V-shaped cleft palates.
-
Eight-year-old boy with Stickler syndrome. Note flat, hypotonic face and small mandible. Patient also has U-shaped, wide cleft palate (CP). His mandible does not show catchup growth. Patient is mouth-breather and snores and is using CP as airway. Closing of CP without preparation would compromise airway passage. Authors recommend placing an obturator (perhaps with speech bulb) for couple of hours daily at first, then gradually increasing time. After few months, when child will have changed breathing pattern, palate can be closed.
-
Eight-year-old boy with Stickler syndrome. Note flat, hypotonic face and small mandible. Mandible does not show catchup growth.
-
Eight-year-old boy with Stickler syndrome. Note U-shaped, wide cleft palate.
-
Pierre Robin sequence in some recognized and unrecognized syndromes.
-
Pierre Robin sequence and complexes.
-
Robin sequences and complexes.
-
Child with Pierre Robin sequence before distraction osteogenesis.
-
Distraction osteogenesis is completed, and bone is consolidating.





