Overview
The backbone of cancer therapy in pediatric oncology has been stepwise integration of multimodality therapies (eg, chemotherapy, surgery, radiation therapy) into carefully designed treatment regimens tested sequentially through multicenter randomized trials. Although chemotherapy has been effective in eradicating micrometastatic disease in some conditions and remains a principal determinant of success, the toxicity associated with current cytotoxics is substantial, and current agents have not proven curative in several clinical groups.
Hence, the need is growing for the development of effective, alternative anticancer therapies for use in children with tumors. Dramatic progress in technology has improved our understanding of the basic biology of tumor immunology, and immune-based therapies represent one approach that could be integrated into current multimodal regimens to eradicate micrometastatic disease.
Immunotherapeutic targeting of cancers is an attractive, novel modality that could be used either in conjunction with conventional therapies or separate from these therapies. Evidence currently suggests that the mechanisms responsible for resistance to cytotoxic agents generally do not confer resistance to immune-mediated mechanisms of tumor-cell killing. [1, 2, 3, 4, 5, 6]
The immune system participates in immunosurveillance of tumors, as evidenced by the following:
-
Patients develop spontaneous innate and acquired immune responses to their tumors. [7]
-
One positive prognostic indicator of patient survival in many histologies is infiltration of lymphocytes within a tumor. [7]
-
Immunosuppressed transplant recipients display higher incidences of nonviral tumors than age-matched immunocompetent controls. [7]
-
Various murine studies have reported that both spontaneous and carcinogen-induced tumors occur more frequently in mice that lack various elements of innate and adaptive immunity. [8]
-
Tumors are less able to survive in immunocompetent mice. [7]
This last observation provides evidence that the development of immune-evasive properties occurs in response to immune pressure early during the period of oncogenesis. Merchant and colleagues also suggest that T-cell–depleting cancer therapies may eliminate beneficial immune responses and that immune reconstitution of patients with lymphopenic cancer could prevent metastatic recurrence of solid tumors. [9]
In summary, current models hold that tumor antigens are present and induce immune reactivity during incipient tumor growth and that tumors subsequently develop properties to evade these immune responses. The challenge to the current field is to elucidate the biology of host-tumor interactions at the time of clinical presentation with cancer and to develop approaches that diminish the capacity for tumors to evade immunity and amplify host antitumor immune responses. In pediatric oncology, the ability to apply such therapies during a period of minimal residual disease is compelling and can often be accomplished with modern multimodality therapies.
The immune system consists of multiple levels of defense against invading pathogens. It consists of physical barriers, mechanisms of innate immunity as well as adaptive immunity. The physical barrier can be mechanical, chemical, or biological and located at different sites in the body including skin, secretions within the respiratory tract, and in the gastrointestinal or genitourinary tract as flora. The cells that have the inherent property of innate and adaptive immunity within the body are present at different sites including the blood, lymphatic system (lymph, lymphoid nodules and lymphoid organs), epithelium, and connective tissues. For more information about the relevant anatomy, see Immune System Anatomy.
Current immunotherapies may involve either innate or adaptive (acquired) immunity.
Innate immunity does not induce immunologic memory. It can directly kill tumor cells. This immunity plays a critical role in initiating adaptive immune responses. Innate immunity includes the role of toll-like receptors (TLRs) in improving antitumor and vaccine responses, muramyl tripeptide phosphatidylethanolamine (MTP-PE) in osteosarcoma, and natural killer (NK) cell–killer immunoglobulinlike receptor (KIR) mismatch. This can also augment tumor growth and invasion.
Adaptive immunity induces immunologic memory and can directly kill tumor cells or recruit other effectors through cytokine production. B cells are professional antigen-presenting cells (APCs) that generate antibodies in response to a foreign antigen.
T cells are potent tumor cell killers that recognize peptides derived from the target cell (extracellular or intracellular) but must be presented by major histocompatibility complex (MHC) molecules. Activation of T cells also requires a second signal provided by a costimulatory molecule. T cells used in a donor lymphocyte infusion or as part of a stem cell transplant can have potent graft-versus-leukemia effects.
Vaccines have been designed as a means of providing a targeted tumor antigen with an appropriate costimulatory signal to enhance T-cell responses. Cytokines and growth factors have been used on pediatric tumors as a means of directly killing tumors or improving host antitumor responses but with limited success.
Monoclonal antibodies (mAbs) target only cell surface antigens and can induce antibody-dependent cellular cytotoxicity (ADCC) or complement fixation; they can be directly cytotoxic or serve as targeting agents to deliver lethal “hits.” mAbs have been used as single agents or in combination with chemotherapy and can be conjugated with radioisotopes or toxins to enhance their potency.
Go to Induction of Tolerance for information on this topic.
The Pediatric Immunotherapy Discovery and Development Network (PI-DDN) is a collaborative network that was created to identify and advance preclinical immunotherapy research for children and adolescents with cancer. [10] Highlights of PI-DDN research include the following:
-
Understanding and overcoming immune cell exhaustion to improve the cancer-fighting ability of CAR T cells [11]
-
Identifying molecules on the surface of cancer cells that can be targets of immune-based therapies. (eg, the use of CAR T cells that are engineered to target the GPC2 protein in pediatric brain tumors) [12]
-
Identifying immune signatures of acute myeloid leukemia that are predictive of chemotherapy and immunotherapy response and resistance [13]
Activation of Innate Immune System
William Coley, MD, a pioneering surgeon who practiced medicine at Memorial Hospital in New York City from 1890 to 1936, is regarded as the father of cancer immunotherapy. [14, 15] In his practice, he focused on the treatment of sarcomas and developed a firm belief that activation of endogenous immune responses could induce remission of tumors. This conviction arose from reports and direct observations of spontaneous tumor remissions that were temporally related to bacterial infections and were primarily observed in patients with sarcoma.
Coley subsequently produced crude bacterial extracts that were eventually termed “Coley toxins” and were administered to patients with cancer. In some cases (albeit relatively infrequently), dramatic antitumor effects were observed. Ewing sarcoma was one of the tumors in which Coley observed antitumor responses using this approach. [14, 16]
During the same period in which Coley was observing dramatic tumor responses with Coley toxins, the spectacular radiosensitivity of Ewing sarcoma was observed by James Ewing, MD, the physician-in-chief at Memorial Hospital. This led to a rivalry between advocates of immunotherapy (in the form of Coley toxins) and advocates of radiotherapy (such as Ewing). [15] For various reasons, not the least of which was difficulty in standardizing the toxins, immunotherapy was eventually abandoned in favor of cytotoxic radiotherapy, a legacy that persists to this day.
Today, bacteria are known to be potent inducers of the body’s first-line defense, the innate immune system. Activation of innate immunity can not only induce direct antitumor effects but also boost adaptive immunity (especially T-cell responses) and assist in improving tumor antigen presentation. [17] As knowledge of the innate immune system has advanced, the possibility of generating a modern Coley toxin that could incorporate specific activators of innate immunity has increased.
Innate immunity refers to cellular components that serve as the first-line immunologic defense that participates in rapid, nonselective protection without resulting immunologic memory. Stimulation of toll-like receptors (TLRs), which recognize highly conserved structural and molecular patterns on pathogens, is critical to initiating activation of antigen-presenting cells (APCs), which have recently been found to be involved in the efficacy of radiation therapy. [18]
Coley toxins likely work through stimulation of TLRs; accordingly, many investigators currently seek to optimize TLR activation as a means for potent, reproducible activation of innate immunity.
Toll-like receptor activation
A total of 13 TLRs have been identified in humans, each binding 1 or more specific TLR ligands. [19] TLRs can be activated by proteins that are released by or associated with pathogens (ie, lipopolysaccharide, double-stranded RNA, double-stranded DNA) or released by proteins exposed by tissue damage. Although pathogen-derived TLR ligands are well characterized, elucidation of TLR ligands derived from self-tissues continues.
The therapeutic potential of TLR-targeted therapies has been realized in 2 adult cancers by using the cell-wall skeleton of bacillus Calmette-Guérin (BCG-CWS), a TLR2 and TLR4 agonist, in the treatment of bladder cancer, [20] and using imiquimod, a TLR7 agonist, in the treatment of basal cell cancer. [21] Clinical trials are ongoing in several other adult malignancies, but preliminary results suggest that these may not be sufficient as single agents to induce regression of established bulky tumors. [21]
Such therapies have not yet made their way into pediatric oncology clinical trials, but this may change. Several animal models of pediatric tumors have responded to CpG oligodeoxynucleotides, a TLR9 agonist, including acute myelogenous leukemia (AML), lymphoma, neuroblastoma (see the image below), and rhabdomyosarcoma. [22] Aluminum salts, an adjuvant with TLR4 agonistic properties, have been engineered into vaccines used in children (eg, human papillomavirus vaccine) to augment immune responses and prevent development of secondary cervical cancer. [21]
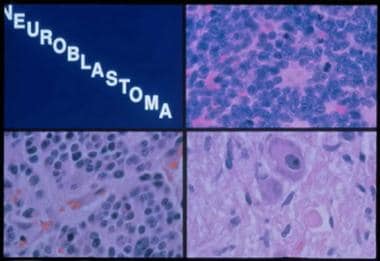 Histologic subtypes of neuroblastoma. Top right panel, neuroblastoma: A monotonous population of hyperchromatic cells with scant cytoplasm. Bottom left panel, ganglioneuroblastoma: Increased schwannian stroma. Bottom right panel, ganglioneuroma: Mature ganglion cell with schwannian stroma.
Histologic subtypes of neuroblastoma. Top right panel, neuroblastoma: A monotonous population of hyperchromatic cells with scant cytoplasm. Bottom left panel, ganglioneuroblastoma: Increased schwannian stroma. Bottom right panel, ganglioneuroma: Mature ganglion cell with schwannian stroma.
Tumor cells can also release or expose damage-associated molecular patterns, which include lipids and lipopeptides, proteins, and nucleic acids that bind to TLRs and cause activation of macrophages and dendritic cells (DCs). [21]
The release of inflammatory mediators by damaged cells also plays a central role in amplifying and directing specific T-cell responses. Recent studies have investigated the contribution of such damage-associated molecular patterns in the antitumor effects seen with more conventional cytotoxic therapies. For instance, deficiency of TLR4 compromises the efficacy of chemotherapy or radiotherapy in vivo. [23]
Moreover, tumor irradiation has been recently hypothesized to activate effectors of innate immunity through the induction of tumor-cell apoptosis and the release of endogenous TLR agonists, such as heat-shock proteins, uric acid, or high-mobility group box protein 1 (HMGB1). Whether this relates solely to release of damage-associated molecular patterns by dying eukaryotic cells or is an indirect effect of mucosal damage with systemic seeding of bacteria remains unclear.
Whole-body irradiation was recently shown to increase bacterial translocation and circulating levels of the TLR4 agonist lipopolysaccharide. [18] This platform has been shown in a preclinical model to enhance the effectiveness of adoptively transferred CD8+ T cells in tumor-bearing, lympho-depleted mice, leading to improved tumor regression, whereas tumor-bearing mice deficient in TLR4 do not benefit from whole body irradiation. [24]
Thus, although radiation may have been traditionally viewed as a purely cytotoxic therapy to shrink tumors, it may also have significant immunomodulatory properties.
Danger signals in rhabdomyosarcoma and other tumors
The release of inflammatory mediators (alarmins) as tumors die off can lead to immune responses against self-molecules. HMGB1 is one such nuclear protein that binds the receptor for advanced glycation end products (RAGE) TLR2 or TLR4. [25, 26] Tumor cells that undergo apoptosis in vitro in response to anthracyclines can be picked up by DCs through TLR4 and lead to long-term tumor protection without addition of adjuvants.
The relevance of this in pediatric oncology has been demonstrated in rhabdomyosarcomas, in which preclinical data have shown that HMGB1 stimulates myogenesis through RAGE; this tumor may reduce expression of RAGE as a means of survival. [27] The implications are that alarmins are a byproduct of tumor cell death, are purposefully underexpressed by tumors to avoid detection by the immune system, and are a potential novel target for immunotherapy.
Muramyl tripeptide phosphatidylethanolamine in osteosarcoma
Muramyl tripeptide phosphatidylethanolamine (MTP-PE) is an analog of muramyl dipeptide, a substance contained within the cell wall of mycobacteria that has immune-activating activity. When MTP-PE is encapsulated in multilamellar liposomes (L-MTP-PE), it is efficiently delivered to the liver, spleen, lung, nasopharynx, and thyroid after intravenous (IV) infusion. [3]
L-MTP-PE can bind to TLR4 on monocytes and macrophages, leading to activation of these cells and promoting antitumor activity. [28] Presumably, the antitumor effects of MTP-PE are mediated via release of interleukin (IL)-1β, IL-6, IL-8, nitric oxide (NO), prostaglandin E2 (PGE2) and tumor necrosis factor alpha (TNF-α). [29] The exact mechanism whereby monocyte/macrophage activation and cytokine release results in tumor cell death remains incompletely understood.
MTP-PE administration has been primarily studied in pediatric patients with osteosarcoma, a disease that frequently metastasizes to the lung and a disease in which micrometastases cause treatment failure in almost 40% of patients despite administration of multiagent chemotherapy.
In a phase II trial, MTP-PE prolonged disease-free survival in a group that received 24 weeks of therapy. [3] In addition, peripheral fibrosis, inflammatory cell infiltration, and neovascularization were observed in metastases from recipients of MTP-PE but not in control subjects. Thus, this trial suggested that L-MTP-PE is an active biologic agent that can produce a survival benefit in this patient population. [29]
Further investigation of MTP-PE in patients with osteosarcoma was undertaken in a phase III trial, in which improved survival rates were noted in patients who received ifosfamide-containing cytotoxic drug therapy (ie, cisplatin, methotrexate, doxorubicin [Adriamycin], and ifosfamide) with MTP-PE but were not observed in those receiving MTP-PE without ifosfamide. [29]
Notably, this study was configured with a factorial design, wherein patients were randomized to 1 of 4 arms, with the intent to compare the use of 3 standard drugs versus 4 standard drugs and, in a separate question, to answer whether the addition of MTP-PE improved outcome. Because no benefit was observed when MTP-PE was used in the absence of ifosfamide, which was an unexpected finding, the investigators were not able to conclude a definitive benefit from MTP-PE as a single agent.
To date, MTP-PE has not been approved by the US Food and Drug Administration (FDA) for use in osteosarcoma. At present, therefore, it is not readily available for treatment of patients with this disease.
Natural killer cell–killer immunoglobulinlike receptor mismatch
Natural killer (NK) cells are lymphoid cells that are part of the innate immune system because they do not express clonotypic receptors or mediate immunologic memory. However, they are potent mediators of antitumor effects because they identify and kill tumor cells in the absence of inflammatory signals and can directly reject tumors.
NK cell activation is a complex process, with interactions among various activating and inhibitory NK cell receptors and various ligands determining whether NK cells activate and kill targets. [30]
The major NK cell inhibitory receptors are the killer immunoglobulinlike receptors (KIRs), which are expressed on the surface and inactivate the NK cell when it encounters a cell that expresses self-major histocompatibility complex (MHC) class I. Activating receptors include NKG2D, which binds MHC class I chain-related molecule A or B (MICA or MICB) on tumor cells, and Fc receptors, which bind the Fc region of antibody molecules.
Several clinical trials have shown that adult patients with AML who underwent bone marrow transplant and were mismatched for NK cell–KIR and MHC displayed improved survival. [31] This success appears to be contingent on using a T-cell–depleted transplant, perhaps because of the absence of immunosuppressive agents that may interfere with the natural killer cell–mediated effects.
A phase II trial through the Children’s Oncology Group is currently exploring the efficacy of allogeneic NK cell–KIR mismatched transplants in children with relapsed, refractory, or newly diagnosed AML, and recruitment is ongoing (see Clinicaltrials.gov).
Limitations of Innate Immune System Activation
Although innate immune effectors can clearly mediate antitumor effects, elements of innate immunity may also contribute to tumor growth. Much recent interest has been focused on the interaction between inflammation and carcinogenesis. Especially in adult carcinomas, chronic inflammation can give rise to cancer, whereas anti-inflammatory therapies can prevent cancer.
Thus, although specific activation of innate effectors (eg, monocytes, macrophages, eosinophils) can induce potent killing of tumor cells, [32, 1, 33, 34] ample evidence also implicates tissue macrophages as contributors to the immunosuppressive microenvironment within tumors themselves and, thus, potential contributors to tumor growth.
M1 versus M2 macrophages
To further the complexity, macrophage differentiation has been characterized as proinflammatory (the M1 variety) and anti-inflammatory (the M2 variety). Tumor-associated macrophages primarily appear to consist of the M2 variety and are known to localize into hypoxic regions of tumors and to secrete various immunosuppressive cytokines. M2 macrophages also promote tumor progression by facilitating angiogenesis and invasion. [35]
In many animal models, macrophage depletion actually results in diminished tumor cell survival, increased tumor rejection, or both. Notably, pediatric tumors show a predominance of macrophage infiltration, suggesting that the immunosuppressive macrophage may have particular importance in pediatric solid tumors. [36]
Type I versus type II natural killer T cells
Natural killer T cells represent another class of cells in which a dichotomy is observed; some subpopulations induce immune activation and antitumor effects, whereas very similar but distinct populations can actually contribute to tumor growth and progression. [37]
For instance, type I (classic) natural killer T cells express the semi-invariant T-cell receptor (TCR) using a unique TCR Vα14Jα18 chain in the mouse and a Vα24Jα18 chain in the human. This TCR recognizes the glycolipid α-galactosylceramide (αGalCer) in association with a nonclassical major histocompatibility complex (MHC) class I molecule called CD1d.
Type I natural killer T cells have been shown to protect against methylcholanthrene-induced sarcomas in vivo, and this effect depends on αGalCer and interleukin (IL)-12 production by dendritic cells (DCs). [38, 39, 40] These cells appear to produce interferon gamma, a cytokine that then recruits natural killer cell and CD8+ T cells to the tumor, leading to lysis.
Type II (nonclassic) natural killer T cells also bind CD1d but lack the Vα24Jα18 chain, have many different TCRs, and, with rare exception, do not recognize αGalCer. This subset is poorly understood but is generally thought to be suppressive of tumor immunosurveillance.
Because of the success of natural killer T cells in eliminating tumors in preclinical mouse models, clinical trials were performed using αGalCer-pulsed DCs and natural killer T cells in patients with lung cancer. Neither administration of the DCs nor adoptive transfer of autologous natural killer T cells activated with αGalCer and IL-2 yielded responses but were well tolerated in phase I trials. [41, 42]
Humans generally have a lower frequency of natural killer T cells, but biologic differences between natural killer T cells in mice and those in humans may explain the disparity; further trials are needed.
Myeloid-derived suppressor cells
The role of myeloid-derived suppressor cells (MDSCs) as an important component of the immunosuppressive microenvironment of tumors is becoming better appreciated. Tumor growth results in expansion of this subset, causing a reduction in arginine levels and a subsequent increase in nitric oxide (NO) in tumors, which inhibits T-cell activation and antigen-specific responses. Therapies that inhibit cyclooxygenase-2 enzyme activity, inhibit MDSC function, or cause MDSC differentiation (eg, all-trans retinoic acid) are being explored in preclinical models. [43]
Activation of Adaptive Immune System
The adaptive (acquired) immune system is composed of T cells and B cells that undergo specific recognition of foreign antigens and generate immunologic memory.
T cells are lymphoid cells that are generated in the bone marrow but undergo maturation in the thymus. They only recognize foreign peptides when presented with a self-major histocompatibility complex (MHC) molecule; this interaction is the first signal needed to activate a T cell. These cells also require stimulation by a second signal, a costimulatory molecule provided by professional antigen-presenting cells (APCs)—typically dendritic cells (DCs)—which instruct T cells to become activated against antigens expressed in the milieu.
In this regard, researchers have designed vaccines that provide these 2 signals to target a tumor-associated antigen. Other than their direct cytotoxic effects against tumors, T cells can also secrete various cytokines that recruit other components of the immune system.
B cells are lymphoid cells that serve as professional APCs but also secrete antibodies in response to a foreign antigen. Generation of monoclonal antibodies against tumor-associated antigens and receptor tyrosine kinases has led to various targeted therapies that have improved tumor responses even in tumors that failed conventional therapies.
T Cell–Based Therapies
Perhaps the most effective form of immunotherapy of cancer is the graft-versus-leukemia effect mediated by allogeneic T cells after bone marrow transplantation for leukemia. [44] The ability of transferred T-cells to eradicate leukemia highly depends on the type of leukemia, the degree of tumor burden, and the rate of disease progression.
Careful clinical trials are necessary to target tumor histologies and tumor burdens most receptive to immune-based therapies, and creative combination strategies are likely to be needed to enhance the effectiveness of T cell–mediated antitumor effects in most clinical settings.
Graft-versus-leukemia effect after bone marrow transplantation
Development of bone marrow transplantation as a clinical approach to malignancy initially rested on the assumption that high doses of chemotherapy were necessary to eradicate relatively chemoresistant leukemia. In this paradigm, the marrow graft served only to rescue marrow function that was irreversibly ablated by high-dose therapy or irradiation.
However, clinical evidence accumulated over the past 25 years has shown that an important component of the graft-versus-leukemia effect in bone marrow transplantation is related to an immunologic reaction that occurs between donor T cells contained in the marrow graft and residual tumor cells that remain after high-dose chemotherapy. [45, 46, 47, 48, 49, 50]
-
Patients who develop some evidence of graft versus host disease (GVHD) experience a lower incidence of leukemic relapse. [51]
Perhaps the most important principle gleaned from the clinical experience is the potency with which T cells can permanently eradicate aggressive, recurrent, and chemoresistant leukemic cells.
Several other lessons are also pertinent. Immune responses can sometimes occur at a relatively slower tempo than that observed with cytotoxic therapies. For example, in CML, detection of molecular evidence of leukemia for at least 6-9 months after bone marrow transplantation is not uncommon, with gradual resolution of molecular evidence of residual leukemia occurring over 1-2 years. [59]
A second important principle is that not all leukemias are equally susceptible to the graft-versus-leukemia effect. For instance, DLIs induce complete responses in 60-80% of patients with stable chronic-phase CML; the response rate is approximately 30% in accelerated-phase CML, and less than 20% of patients in CML blast crisis respond. [57] However, DLIs have had less promising results in other leukemias, including 15-29% response rates in AML and less than 15% in acute lymphoblastic leukemia (ALL). [60]
Thus, polyclonal T cells mediate antitumor responses with a wide range of efficacy, even within a given tumor. The reasons for the differences in susceptibility are not well understood but no doubt hold important clues to the understanding of immune-based mechanisms of antitumor activity.
A growing body of literature suggests that T cells can also exert antitumor effects in some solid tumors, including a 25% response rate in renal cell carcinoma. [61, 62, 63] Whether similar activity is observed against pediatric solid tumors is unknown, but a few institutions have begun trials, ranging from adoptive immunotherapy of vaccine-primed T cells after chemotherapy to allogeneic stem cell transplants for pediatric patients with solid tumors. [64, 65, 66, 67, 68]
New approaches in bone marrow transplantation are also focusing on ways to diminish the toxicity of bone marrow transplantation by reducing the myelosuppression of the preparative regimen while preserving the antileukemic effect that is mediated by T cells contained within the graft. [69] Thus, bone marrow transplantation appears to be evolving away from high-dose myelosuppression and toward adoptive immunotherapy, thereby creating an environment that can enhance the immune-mediated effects.
Tumor vaccines
The efficacy of graft-versus-leukemia in eradicating leukemia illustrates the potential of T cells as effectors of antitumor activity in allogeneic settings. To exploit T-cell reactivity to autologous malignancies, existent weak tumor–directed immune responses must be amplified to result in meaningful antitumor effects. This is particularly challenging for cancer because tumor antigens frequently represent overexpressed self-antigen, for which the quality and quantity of antigen-reactive T cells is limiting.
Therapeutic tumor vaccination is a strategy that initiates a dynamic process of activating the host’s own immune system against a tumor antigen. The immunologic tolerance induced by tumor antigens during the course of primary tumor growth is not absolute and can be overcome through specific immunization with professional APCs or by transducing tumor cells with cytokine or costimulatory receptor-encoding genes. [70]
Although tumor vaccines alone do not induce clinically meaningful antitumor effects in patients with measurable tumor burdens, the usefulness of such immune-based therapies is likely to be optimal in patients with minimal residual disease. [71, 72]
Randomized studies comparing outcomes following tumor vaccination for patients with a high risk of disease recurrence have only rarely been conducted.
Of the 9 vaccines tested in phase III trials thus far, only 1 showed a reduction (31%) in the hazard rate for progression in asymptomatic, metastatic, hormone-refractory prostate cancer: sipuleucel-T (Provenge), which is composed of autologous APCs that express prostatic acid phosphatase and granulocyte-macrophage colony-stimulating factor (GM-CSF). [73] The trial was not designed to look at overall survival as a primary endpoint, but the group who received sipuleucel-T had a 4.5-month increase in overall survival. [73]
Limitations of Adaptive Immune System Activation
Various challenges face the development of immune-based therapies for childhood cancer. One challenge to the development of vaccines for pediatric tumors is the rarity of these tumors; hence, effective testing of vaccine strategies directed at common immunodominant antigens is difficult.
Although identification of a particular peptide that is capable of inducing antitumor effects may be possible in patients with a given major histocompatibility complex (MHC) and pediatric tumor, this is likely to occur in only a few patients per year. Thus, the ability to test such complicated and specific therapies in rare tumors is limited, and the clinical applicability of using specific targeted epitopes is small.
Hence, enthusiasm is greater for targeting more public tumor antigens that are shared across different tumor types and for developing immunizing approaches that can be used across human leukocyte antigen (HLA) types, such as whole-protein or whole-cell techniques.
A second challenge is that because dose-intensive chemotherapy is a standard component of almost all therapies for pediatric tumors, integrating T-cell–based therapies into existing therapies requires attention to the important changes in T-cell homeostasis induced by lymphopenia, which impacts the effectiveness of T-cell–based immunotherapy.
Although pediatric patients have more rapid recovery of immunity after T-cell–depleting chemotherapy than adults do, they still usually need at least 6-12 months to achieve immune recovery after dose-intensive chemotherapy. [74, 75, 76, 77, 78, 79]
Recent studies have demonstrated that lymphopenia can provide a fertile milieu for immunotherapy; however, this is not universally true. In general, lymphopenic hosts appear to provide a reasonably fertile environment for adoptive immunotherapy. Immunization undertaken in the context of lymphopenia can be more effective than when undertaken in T-cell–replete hosts if adequate T-cell numbers and effective antigen-presenting cells (APCs) are provided. [80]
Results of a study of patients with Ewing sarcoma and alveolar rhabdomyosarcoma were recently reported. Adoptive T-cell transfer was administered, with peptide-pulsed dendritic cell (DC) vaccines undertaken during the period of lymphopenia that immediately followed dose-intensive chemotherapy, and the overall survival for patients with metastatic or recurrent Ewing sarcoma or alveolar rhabdomyosarcoma treated with adoptive T-cell transfer and DC vaccination was 43% at 5 years. [78]
Interestingly, although the patients showed minimal measurable immune responses to the peptide-based vaccine, which targeted the translocation breakpoint junctions, they were immunocompetent to influenza vaccination within 3 months following completion of chemotherapy.
A third challenge is that the optimal technique for tumor vaccination has not been defined. Peptide-based vaccines have been successfully used by some groups and are known to be simple to produce, relatively inexpensive, and safe. [81] However, as noted (see above), such an approach requires identification of a tumor-specific or tumor-associated peptide, which is challenging because of the rarity of pediatric tumors.
For neuroblastoma, peptides derived from the amplification of the MYCN oncogene have been shown to be capable of inducing immune responses ex vivo and capable of lysing neuroblastoma cells in patients with HLA A1+ tumors; thus, this target could potentially be clinically exploited in future studies. [82] Peptide-based DC vaccines were developed to target the translocation breakpoints in Ewing sarcoma and alveolar rhabdomyosarcoma, but no clinical responses were observed. [83]
WT1 is a zinc finger transcription factor that is overexpressed in various hematologic and solid tumors, and peptide vaccines that express WT1 have yielded clinical responses in various adult malignancies. [84] It has recently been found to be a target antigen for the graft-versus-leukemia effect. [85] Trials are now underway using vaccines that express WT1 in children (see Clinicaltrials.gov).
An alternative approach is to use components of the tumor cells themselves to potentially immunize patients of any HLA type to various potential antigens available within the tumor. For example, Geiger et al used tumor lysate–pulsed DCs to immunize patients with pediatric solid tumors and reported some preliminary evidence for antitumor effects. [86, 65]
Neuroblastoma cells have also been transduced using complementary DNA-encoding cytokines and have been subsequently used to immunize patients. [87, 88] The rationale for this approach is based on data demonstrating that stimulatory cytokines can generate immune responses that show cytolytic activity for both cytokine-secreting and non–cytokine-secreting cells that bear the same antigens.
Clinical evidence for antitumor activity was obtained by using autologous neuroblastoma cells transduced to secrete interleukin (IL)-2, which resulted in a 20% objective tumor response and a 30% stable disease rate in 10 patients. [89] As a result of the technical difficulties of reliably generating gene-transfected tumors in patients, these investigators have also explored the use of allogeneic neuroblastoma cells transduced with IL-2. [87] Neither treatment produced any toxicity aside from local induration at the site of tumor injection.
Repeat studies attempted to improve the outcome of this approach by cotransfecting the complementary DNA–encoding cytokines with the chemokine lymphotactin in autologous neuroblastoma cells; a few complete and partial responses were noted in one trial, but no responses were noted in the other. [90, 91]
The potential of using master cell banks of allogeneic tumors as immunogens is compelling because of the ability to generate an “off-the-shelf” reagent that can be applied across patients. Whether such allogeneic tumor cells lines provide a more effective or less effective immunogen remains to be determined.
Cytokine and Growth Factor Therapy
Future studies will seek to combine T-cell–based vaccination protocols with other modalities to amplify the magnitude of the immune response. For instance, various adjuvants, such as granulocyte-monocyte colony-stimulating factor (GM-CSF) and toll-like receptors (TLRs), can be used to better activate antigen-presenting cells (APCs); these can potentially result in more effectively activated or expanded T cells.
Alternatively, cytokines, growth factors, and costimulatory molecules that help in the expansion and activation of T cells are also under investigation. For instance, both interleukin (IL)-7 and IL-15 are γ-c signaling cytokines that substantially augment T-cell response to vaccination and have been shown to improve antitumor effects in animal models. [92, 93] Costimulatory molecules include 4-1BB ligands, OX40 ligands, and CD28 ligands, all of which can enhance T-cell expansion after activation. Anti-4-1BB antibodies have been shown to have potent antitumor effects in vivo.
Finally, inhibition of regulatory pathways remains a major area of study because suppressive mediators released by tumors play a central role in limiting antitumor immunity. Among these are blocking antibodies to CTLA4, anti-CD25 to deplete regulatory T cells, and blockers of PD-1 and its ligand, PD-L1. The combination of these strategies that will ultimately be most promising in augmenting antitumor immunity remains to be seen.
IL-2 is active against renal cell carcinoma and malignant melanoma; however, for pediatric tumors, several trials of IL-2 have been performed with no antitumor effects observed. [94, 95, 96] Even in neuroblastoma, a potentially immunoresponsive solid tumor, [97, 98, 99] systemic administration of IL-2 as a single agent has shown no benefit. [95]
Moreover, recent studies have clearly demonstrated that in addition to activating natural killer (NK) cells (the presumed mechanism responsible for the antitumor effects), IL-2 also substantially expands and activates CD25+ CD4+ regulatory T cells. [100] This subset is a significant mediator of tumor-induced immune suppression; therefore, the administration of IL-2 in the context of cancer is likely to amplify this already suppressive subset.
Few other cytokines have been administered as single agents in children with cancer. Interferon alfa (IFN-α) is probably the most well-studied cytokine and has been documented in the treatment of patients with chronic myelogenous leukemia (CML), hairy cell leukemia, some B-cell and T-cell lymphomas, and solid tumors, such as melanoma, renal carcinoma, and Kaposi sarcoma, all of which are very rare in pediatric patients.
IFN-α is approved by the US Food and Drug Administration (FDA) for the adjuvant therapy of adults with stage III melanoma. An ongoing clinical trial is examining the use of IFN-α in children with melanoma. [101] Ongoing phase I trials are also exploring the role of pegylated IFN-α for plexiform neurofibromas and brain tumors in children (see Clinicaltrials.gov). How IFN-α mediates antitumor effects remains unclear.
Regional therapy with tumor necrosis factor alpha (TNF-α) has been performed in patients with sarcoma, and some antitumor responses were observed in Ewing sarcoma and Wilms tumor, although this approach is limited by the development of systemic toxicity. [102]
Recombinant GM-CSF (sargramostim) is a growth factor that may act on cancer cells through a role in cytokine priming. When given before and during induction chemotherapy, it may make leukemic blast cells more susceptible to the cytotoxic effects of chemotherapy. This growth factor causes upregulation of costimulatory molecule expression on leukemia blasts in vitro and, in combination with INF-α, can induce antitumor immune responses in relapsed acute myeloid leukemia (AML) and acute lymphoblastic leukemia (ALL) after allogeneic stem cell transplantation. [103]
Tumor cells engineered to secrete GM-CSF are particularly effective as antitumor vaccines, and the addition of GM-CSF to standard vaccines may increase their effectiveness by recruiting dendritic cells (DCs) to the site of vaccination. [104] GM-CSF is also commonly administered locally at the site of administration of tumor vaccines, where it presumably enhances antigen presentation via recruitment of antigen presenting cells to the site where the immunogen is deposited.
A novel application of inhaled GM-CSF may be for children with pulmonary metastases from sarcomas, with 1 child with Ewing sarcoma demonstrating a complete response. [105, 106] Of 40 patients treated with pulmonary metastases, 24 had disease stabilization or partial regression for a mean duration of 10 months. [106] This included 8 of 13 patients with sarcoma who responded.
Monoclonal Antibody Therapy
When Kohler and Milstein reported the technology for generating monoclonal antibodies (mAbs) in 1975, many tumor biologists expected the rapid evolution of various antibodies that could act as “magic bullets” to target and kill tumors. [107] Although progress in the field of mAb therapy for neoplasia has proceeded much slower than was initially anticipated, recent clinical trials have demonstrated antitumor activity in various malignancies.
Effective mAb therapy for cancer requires the identification of appropriate tumor-specific targets expressed on the surface of the cancer cells. For effective targeting, the molecule should be relatively tumor-specific (eg, show substantially less binding to normal tissues or binding to only “dispensable tissue” (eg, B cells) and should be highly expressed in tumors (compared with normal tissues).
In addition, the target of the mAb should not be shed from the tumor following mAb binding; rather, the antigen-mAb complex should be internalized by the tumor cell. Importantly, many antigens are down-regulated after antibody binding, which can limit the effectiveness of the therapy.
Furthermore, because mAbs are theoretically “foreign proteins” and therefore could induce an immune response that might limit their efficacy, they must be rendered sufficiently nonimmunogenic to prevent development of neutralizing antibodies.
mAbs were initially produced by fusing murine myeloma cells with B cells from mice immunized with specific antigens. Because mAbs generated in this manner are murine proteins, they are recognized as foreign in immunocompetent humans and, thus, generate neutralizing antimurine antibodies termed human antimouse antibodies (HAMAs). After development of these antibodies in patients, the half-life of mAbs is greatly reduced, which significantly reduces their biologic activity.
Advances in genetic engineering subsequently allowed the creation of chimeric antibodies, which use human constant regions and retain the entire murine variable region, and humanized antibodies, which retain only the murine hypervariable region responsible for epitope binding. The rest of the variable region and the entire constant region are human derived. More recently, fully human mAbs have become readily available and further limit the likelihood of neutralizing antibody development.
The overall success of mAb therapy in cancer is determined by the ability of antibody binding to result in tumor cell death. This can occur via 1 of the following 3 pathways, which are not mutually exclusive:
-
Immune effects through antibody-dependent cellular cytotoxicity (ADCC) and complement-mediated cytolysis - Binding the antibody recruits cells with Fc receptors to the site of the tumor, which then kill the tumor cell, or complement is fixed and the tumor cell is killed.
-
Direct killing - Key process include interruption of a critical cell signaling cascade by inhibition of ligand binding; downregulation of a receptor tyrosine kinase, which transmits a necessary life signal; and induction of an apoptotic signal following ligation of the target by the mAb.
-
Targeting via a conjugated antibody of antibody receptor (eg, radionuclide, immunotoxin, cell-based genetic fusion) - This targets a lethal “hit” to the tumor cell.
Ongoing work in this field is focused on identifying approaches to increase ADCC by using agents such as granulocyte-macrophage colony-stimulating factor (GM-CSF) to improve recruitment of effector cells in combination with an mAb for refractory osteosarcoma and neuroblastoma. [108, 109, 110] Indeed, differences in the relative potency of ADCC effects with differing antibodies [111] and with genetic variation appear to be a primary determining factor in whether mAb therapy for cancer is effective. [32]
ADCC appears to be the primary mechanism of action for mAbs targeting the GD2 disialoganglioside in neuroblastoma. Studies have shown that neuroblastoma is susceptible to ADCC via lymphocytes, neutrophils, and activated macrophages. GD2 is expressed at high densities on nearly all neuroblastoma cells, is not shed from the cell surface, and is restricted to neuroectodermal tissues, thus representing a good potential target for mAb therapy. [112] Several antibodies directed against GD2 on the surface of neuroblastoma cells have been developed.
Initial studies using 3F8, an anti-GD2 mAb, demonstrated that the primary adverse effects of therapy were limited to results of acute toxicity. [113] Despite toxicity, 3F8 mAb can be administered in the outpatient setting with symptomatic management of the toxic effects. Results of initial nonrandomized clinical trials report a long-term disease-free survival rate in patients with stage IV neuroblastoma that is comparable to the rate in historical control subjects without mAb exposure. [113]
Interestingly, evidence suggests that low levels of HAMAs and the development of nonneutralizing anti-idiotypic antibodies (antibodies directed to the variable region of the immunizing antibody rather than to the constant region, which is the target of most HAMAs) correlate with improved survival rates after adjuvant 3F8 mAb therapy. [114] Trials using another anti-GD2 mAb, hu14.18, linked with either interleukin (IL)-2 or GM-CSF, also had toxicity that was reversible, but no clinical responses were documented. [115, 109]
mAb therapy may also effectively kill tumors when the mAb induces death in tumor cells after antibody binding. [116] Examples of this approach include the anti-CD20 mAb rituximab in lymphoma [117] and anti-CD99 in Ewing sarcoma. [118, 119] Unfortunately, CD99 is also expressed on hematopoietic progenitors and T cells, thus limiting its clinical potential for tumor targeting.
In pediatric studies, mAbs targeting vascular endothelial growth factor (VEGF) as well as the immunoglobulin (Ig)F-1 receptor have shown progress in preliminary reports, presumably via interruption of signaling pathways critical for tumor survival. [120, 121]
Nonconjugated antibodies can also induce cell death if they crosslink a cell surface receptor that can initiate a downstream death cascade. Antibodies to the tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) receptors show significant activity in vitro in pediatric tumors by initiating such a death pathway. [122] Antibodies to CD40 can induce similar effects in melanoma and some carcinomas. [123, 124]
Recent evidence has suggested that the capacity to induce cell death as a result of mAb binding can be synergistic with cytotoxic chemotherapy. Whether this synergism results from enhanced ADCC, from enhanced effects of an interrupted growth signal, or from both is not clear in every case. One example of this is trastuzumab, which targets the HER2 or NEU protooncogene in breast cancer and has substantially increased the chemotherapy response rate in various clinical trials. [125]
On the basis of the association between HER2 expression and worse survival in osteosarcoma, [126] trastuzumab has recently been used in conjunction with standard chemotherapy for HER2 -positive osteosarcoma. However, a concern with this approach is that the HER2 expression in osteosarcoma does not result from gene amplification, as it does in breast carcinoma; therefore, interrupting HER2 signaling is not likely to result in death in this tumor.
A third mechanism by which antibodies can kill tumor cells involves targeted delivery of a lethal agent, such as a toxin or radionuclide. With rituximab, an unconjugated mAb against CD20, a 96% overall response rate was reported in one phase II trial for lymphocyte-predominant Hodgkin lymphoma, with 75% remaining in remission after one year. [127] Another phase II trial showed similar results. [128]
A phase II pilot study is underway through the Children’s Oncology Group to assess the toxicity of adding rituximab to upfront chemotherapy for B-cell leukemia and lymphoma (see Clinicaltrials.gov).
Conjugating anti-CD20 to radionuclides has shown success in children with Hodgkin lymphoma, although the bone marrow toxicity is substantial; therefore, this approach is generally undertaken in the setting of marrow rescue. [129] Other mAbs that have been conjugated to toxins in pediatric oncology include anti-CD25 and anti-CD30, conjugated with ricin for Hodgkin lymphoma, [130, 131] and anti-CD22, conjugated with pseudomonal exotoxin for acute lymphoblastic leukemia (ALL). [132]
In pediatric acute myeloid leukemia (AML), the differentiation antigen CD33 is expressed in almost all patients. The US Food and Drug Administration (FDA) approved anti-CD33 conjugated with calicheamicin (gemtuzumab [Mylotarg]) for treatment of adult AML in 2000, but the agent was withdrawn from the US market on June 21, 2010.
Apart from some infusional allergic reactions, the primary toxicity of this approach has been bone marrow suppression caused by binding the mAb-toxin conjugate to normal hematopoietic precursors that express CD33. Another still unexplained toxicity of anti-CD33–calicheamicin conjugates is hepatic damage, which is characterized by transient increases in liver enzymes in approximately 25% of patients and, occasionally, a more severe complication consistent with veno-occlusive disease. [133]
Currently, the agent is being tested both as a single agent and in combination with chemotherapy in children with AML; however, results of phase I and II clinical trials indicate promise, with an overall remission response rate of 45% and a 1-year event-free survival and overall survival estimates of 38% and 53%, respectively. [134, 135]
Genetic engineering has recently opened serious prospects of using the cytolytic machinery of T cells or natural killer cells with the effective targeting properties of antibodies via creation of so-called chimeric antigen receptors (CARs). In this case, a single-chain Fv-Fc fragment of an mAb is fused to the signaling chain of the T-cell receptor and is introduced genetically into T cells or natural killer cells.
The cells are then systemically delivered or delivered at the site of the tumor; following engagement of the chimeric receptor with its target, T-cell activation or natural killer cell activation leads to tumor cytolysis. Promising preclinical results have been recently reported using direct administration of such T cells expressing CARs in a murine model of medulloblastoma. [136]
Questions & Answers
Overview
What is immunotherapeutic targeting of cancers in pediatric oncology?
What is the pathophysiology of immunotherapeutic targeting of pediatric cancers?
What is the role of HMGB1 in immunotherapeutic targeting in pediatric oncology?
What is the role of MTP-PE in immunotherapeutic targeting in pediatric oncology?
What is the role of NK cell activation in immunotherapeutic targeting in pediatric oncology?
How do macrophages affect immunotherapeutic targeting in pediatric oncology?
How do MDSCs affect immunotherapeutic targeting in pediatric oncology?
What are the T-cell-based immunotherapeutic targets in pediatric oncology?
What is the graft-versus-leukemia effect after bone marrow transplantation?
What is therapeutic tumor vaccination?
What is the role of monoclonal antibodies in immunotherapeutic targeting in pediatric oncology?
-
Histologic subtypes of neuroblastoma. Top right panel, neuroblastoma: A monotonous population of hyperchromatic cells with scant cytoplasm. Bottom left panel, ganglioneuroblastoma: Increased schwannian stroma. Bottom right panel, ganglioneuroma: Mature ganglion cell with schwannian stroma.
Tables

What would you like to print?
- Overview
- Activation of Innate Immune System
- Limitations of Innate Immune System Activation
- Activation of Adaptive Immune System
- T Cell–Based Therapies
- Limitations of Adaptive Immune System Activation
- Cytokine and Growth Factor Therapy
- Monoclonal Antibody Therapy
- Questions & Answers
- Show All
- References





