Practice Essentials
Deficiency of biotin, a water-soluble B vitamin, may occur from nutritional causes, but more commonly results from deficiencies of enzymes involved in biotin homeostasis (eg, biotinidase deficiency). Affected patients can present with abnormal skin and hair changes, and metabolic and neurologic abnormalities. In severe cases, without treatment, coma and death may ensue.
Early detection and treatment with pharmacologic doses of biotin are important to prevent the development of irreversible complications. Marginal biotin deficiency has been demonstrated in pregnancy and lactation, but the clinical significance is uncertain. [1] Biotin supplements have been promoted to improve skin, hair, and nail health; however, robust evidence for their efficacy is lacking. It is important to be aware that intake of biotin supplements may lead to interference with certain laboratory tests, leading to false-positive or false-negative results. [2] High-dose biotin has been found to be helpful in certain neurologic conditions (eg, multiple sclerosis); however, the mechanism of action is uncertain
Background
Biotin or B7, one of the B vitamins, is an essential nutrient that plays key roles in the metabolism of glucose, amino acids, and fatty acids. Studies support a role for biotin in cell proliferation, DNA repair, and epigenetic gene regulation, as well as normal immune function. [3, 4, 5]
Biotin is available from various food sources and is also synthesized by intestinal bacteria; hence, dietary deficiency is uncommon. Persons with chronic alcoholism and those on long-term anticonvulsants could develop biotin deficiency because of impaired intestinal uptake of the vitamin. Avidin, a protein found in egg whites, binds strongly to biotin, impairing the absorption of the vitamin, leading to severe biotin deficiency in those who consume excessive amounts of raw eggs. Patients affected by certain genetic defects affecting biotin metabolism present with a clinical picture of biotin deficiency.
The clinical presentation of biotin deficiency involves abnormalities of the hair, skin, nails, and the central nervous system. Seizures, hypotonia, ataxia, optic atrophy, visual deficits, sensorineural deafness, and developmental delay (in children) are some of the neurologic manifestations. Metabolic abnormalities may include organic aciduria, lactic acidosis, and hyperammonemia. Supplementation with biotin leads to clinical improvement in most cases.
Pathophysiology
History
In the early 1900s, researchers found that the inclusion of large amounts of raw egg whites in diets in rats produced symptoms of toxicity. In 1926, Boas referred to these symptoms of toxicity as egg-white injury syndrome. [6] The major findings included severe dermatitis, loss of hair, and lack of muscular coordination. Boas also noted that yeast, liver, and several other foodstuffs contained a substance that protected rats from egg-white injury syndrome. A search for this protective factor led to the discovery in 1936 of biotin.
The biochemical basis for egg-white injury syndrome was quickly elucidated when raw egg whites were found to contain the glycoprotein avidin, which has a remarkable affinity for biotin. The biotin-avidin bond is essentially irreversible; as a result, biotin is not liberated from food, and the biotin-avidin complex is lost in the feces. The final step in solving the mystery of egg-white injury syndrome was the demonstration that the syndrome could be prevented by heating the egg whites, a process that denatures avidin and destroys its affinity for biotin.
Structure
Biotin is a bicyclic molecule composed of a ureido ring fused with a tetrahydrothiophene ring.
The ureido ring is involved in the high affinity binding of biotin to avidin, a glycoprotein found in egg-white. A valeric acid substituent is attached to one of the 2 carbon atoms of the tetrahydrothiophene ring. Through this carboxyl group, biotin is linked covalently to the β-amino group of lysine in 5 carboxylases that play critical roles in intermediary metabolism.
Functions of biotin
In addition to biotin's well-known role as a cofactor in carboxylation reactions, recent studies have shown that biotin plays important roles in the regulation of gene expression and immune function. [7, 4, 3, 8]
Biotin-dependent carboxylation reactions
Biotin functions as a coenzyme in carboxylation reactions involving lipid, glucose and amino acid metabolism. There are 5 biotin-dependent carboxylases each of which exists as an inactive apoform. [5] The enzyme holocarboxylase synthetase (HCLS ) catalyzes the addition of biotin (biotinylation) to the inactive apoform, which leads to the formation of the active carboxylase. In all 5 carboxylases, biotin functions as a coenzyme or prosthetic group that serves as a carrier for CO2 in a multistep reaction.
The five biotin-dependent carboxylases [3] and their functions [4] are described briefly below:
Pyruvate carboxylase (PC) catalyzes the formation of oxaloacetate from pyruvate, a step important in the TCA cycle, gluconeogenesis and lipogenesis; lack of this function can lead to hypoglycemia, ketosis, and lactic acidosis.
Propionyl-CoA carboxylase (PCC) catalyzes the conversion of propionyl CoA to methylmalonyl CoA, which in turn isomerizes to succinyl CoA, and enters the TCA (Kreb's) cycle. PCC is important in the metabolism of odd-chain fatty acids, and the amino acids isoleucine, valine, methionine, and threonine. Lack of this enzyme function can lead to propionic acidemia. Levels of PCC in lymphocytes is a sensitive indicator of biotin status.
3-Methylcrotonoyl-CoA carboxylase (MCC) is involved in the catabolism of the branched-chain amino acid, leucine. Lack of biotin can lead to shunting of leucine catabolism products into an alternative catabolic pathway leading to the production of 3-hydoxyisovaleric acid which is then excreted in the urine.
Acetyl-CoA carboxylase I (ACC I) catalyzes the conversion of acetyl CoA to malonyl CoA, in the cytosol, a step important in lipid synthesis.
Acetyl-CoA carboxylase II (ACC II), catalyzes an identical reaction in the mitochondria; the resultant malonyl CoA plays a regulatory role in fatty acid oxidation.
ACC I is a cytosolic enzyme; the remaining carboxylases are found in the mitochondria.
Clinical manifestations of biotin deficiency can also occur as a result of genetic disorders causing a deficiency of the enzyme holocarboxylase synthetase or deficiencies of the individual carboxylase enzymes.
Gene expression
Studies have shown that biotinylation of histones may play a role in gene expression. Multiple sites that bind to biotin have been identified in human histones. Biotin may also affect gene expression by other mechanisms. A few thousand biotin-dependent genes are known in human cells. Some of the genes influenced by biotin include those encoding for enzymes involved in glucose metabolism (e.g. glucokinase), cytokines like interleukin-2 and the insulin receptor. [9, 3, 4, 5]
Immune function
Studies suggest a role for biotin in antibody production, macrophage function, differentiation of T and B lymphocytes, as well the normal function of natural killer cells. [8] Recurrent infections, especially fungal, are common in patients with biotin deficiency.
Role of biotin in high-dose biotin-responsive neurologic conditions
Biotin-thiamin-responsive basal ganglia disease (BTBGD) is a rare neurological condition which can present with seizures and encephalopathy progressing to coma and death. High-dose biotin ( 5-10 mg/kg/day) has been used to successfully treat this condition, but the mechanism of action is unknown. [3]
High-dose biotin treatment (100-300 mg/day) has been found to improve symptoms in a subset of patients with multiple sclerosis. It is thought that improvement of neurological symptoms may involve improved myelin production secondary to the effect of high-dose biotin on the synthesis of long-chain fatty acids. [3] [10, 11]
Role of biotin in individuals with hair, skin and nail disorders
Biotin supplements are widely used by those hoping to achieve healthier hair, skin, and nails. However, there is limited evidence of the efficacy of biotin for this use. A case-control study by Abdel Rahman et al found no significant difference in serum biotin levels between patients with telogen effluvium and those who did not have this form of nonscarring alopecia. [12]
Biotin in high doses has been found to be helpful in two rare conditions: familial uncombable hair syndrome and brittle nail syndrome. [13] In a study of 541 females presenting with hair loss, serum biotin levels were found to be low in 38% of patients. The author concluded that the etiology of hair loss is multifactorial and biotin supplements may be considered if biotin deficiency has been demonstrated and other causes have been ruled out. [14]
Sources of biotin
Biotin is present in a wide variety of foods (meats, dairy, vegetables, seeds, and nuts) and is also produced by intestinal bacteria. In addition, a substantial proportion of individuals may consume biotin containing dietary supplements. [15]
Biotin physiology
Ingested biotin is present in free and protein-bound forms. Protein-bound forms are digested by gastrointestinal proteases and peptidases to form biocytin and biotin-oligopeptides. Free biotin is released from biocytin and biotin-oligopeptides by the action of intestinal biotinidase. Free biotin is then absorbed in the small intestine via a Na+ dependent, carrier-mediated mechanism, which also transports two other nutrients, pantothenic acid and lipoate and hence is known as the sodium-dependent multivitamin transporter (SMVT). The human SMVT gene is located on chromosome 2p23. SMVT activity is regulated by biotin levels; with it being up-regulated in biotin deficiency and down-regulated with biotin over-supplementation. Bacterially synthesized biotin is present in the unbound form and is absorbed in the large intestine by a similar carrier-mediated mechanism. [4] The combined daily output of biotin in the urine and stool exceeds the dietary intake of biotin, suggesting the important role played by intestinal flora as a source of biotin.
Once absorbed, biotin becomes available for various biotinylation processes. Biotin binds to each of the 5 apocarboxylases to form the corresponding holocarboxylase (see the image below) via the action of the enzyme holocarboxylase synthetase.
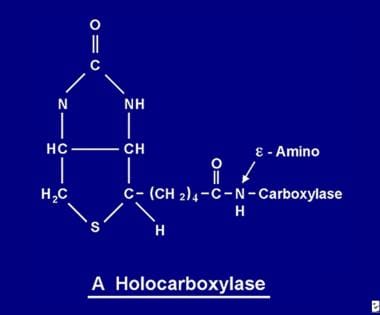 The biotin molecule is bound to the protein by a peptide bond to an e-amino group of an apocarboxylase to form a holocarboxylase.
The biotin molecule is bound to the protein by a peptide bond to an e-amino group of an apocarboxylase to form a holocarboxylase.
Recycling of biotin
After the holocarboxylase enzyme has performed several carboxylations it is captured by cellular lysosomes. In the lysosomes, various proteolytic enzymes degrade the holocarboxylase to form biocytin, which, in turn, is hydrolyzed by the enzyme biotinidase to form biotin and lysine. Free biotin is then available for insertion into an apocarboxylase to form a new holocarboxylase molecule. This recycling process is not 100% efficient. As a result, small amounts of free biotin (and some biocytin) escape the cycle and are lost in the feces and urine. For this reason, biotin must be supplied to the intestine, to replenish the biotin lost from the body. The steps involved in the recycling of biotin- its entry into the gut, its absorption, its incorporation into holocarboxylases, which in turn are broken down to liberate free biotin constitute the biotin cycle and is depicted in the image below.
The enzyme biotinidase is essential for biotin recycling and individuals with biotinidase deficiency will therefore present with signs and symptoms of biotin deficiency.
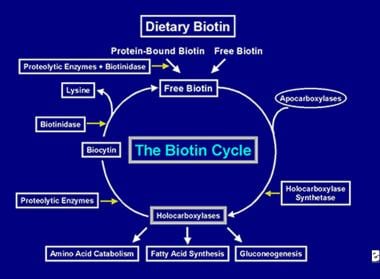
Causes of biotin deficiency
As mentioned earlier, biotin is widely available in foods, is also produced by intestinal flora and is extensively recycled in the body with the help of the enzyme biotinidase; hence biotin deficiency in healthy individuals eating a normal diet is rare. Conditions that can lead to biotin deficiency are described below:
Excessive consumption of raw egg whites: Avidin in raw egg whites has a high affinity for biotin, making it unavailable for absorption. Heat destroys the avidin, so those who eat cooked eggs are not at risk of biotin deficiency. Raw egg whites lead to biotin deficiency only when eaten in excessive amounts (perhaps a dozen or more a day). [16]
Total parenteral nutrition without biotin supplementation: Several cases of biotin deficiency in patients receiving prolonged total parenteral nutrition (TPN) therapy without added biotin have been reported. [17] Therefore, all patients receiving TPN must also receive biotin at the recommended daily dose, especially if TPN therapy is expected to last more than 1 week. All hospital pharmacies currently include biotin in TPN preparations. [18]
Use of infant formulas with inadequate biotin: Biotin deficiency has been reported in infants receiving hypoallergenic formulas. [19]
Chronic anticonvulsant therapy: Prolonged use of the anticonvulsants, phenobarbital, phenytoin, primidone, and carbamazepine, have been linked to biotin deficiency. Possible mechanisms include inhibition of biotin uptake across the intestinal mucosa, accelerated biotin catabolism and impaired renal reabsorption of biotin. Therefore, supplemental biotin has been suggested for patients who are treated with anticonvulsants that have been linked to biotin deficiency. [4, 20]
Prolonged oral antibiotic therapy: Prolonged use of oral antibiotics has been associated with biotin deficiency. Inhibition of intestinal flora that produces biotin is presumed to be the basis for biotin deficiency. Another possible mechanism could be antibiotic-induced overgrowth of bacteria that consume biotin. [21]
Smoking and chronic alcoholism: Studies have shown that smoking may accelerate biotin catabolism, especially in women. Chronic alcoholism may cause intestinal malabsorption of biotin. [4, 22]
Short gut syndrome and inflammatory bowel disease: Individuals with short bowel syndrome and inflammatory bowel disease are also at risk of biotin deficiency as a result of intestinal malabsorption of biotin.
Marginal biotin deficiency during pregnancy and lactation: Recent studies have shown decreased biotin levels in a significant proportion of pregnant and lactating women. There are concerns that marginal biotin deficiency during pregnancy might be teratogenic and some experts have recommended a higher intake of biotin by pregnant women. [3]
Certain inborn errors of biotin metabolism may also lead to the manifestation of biotin deficiency.
Biotinidase deficiency (BTD) is inherited in an autosomal recessive fashion and occurs at a frequency of approximately 1 in 60,000 live births.; an estimated 1 in 120 individuals are heterozygous for the condition. In individuals homozygous for the disorder, biotinidase levels are < 30% normal leading to biotin deficiency from insufficient free biotin release due to diminished biotin recycling. Symptoms of BTD typically develop between 1 week and 1 year of age. The severity of the enzyme deficiency may vary. Those with profound biotinidase deficiency have BTD levels less than 10% normal, while those with partial biotinidase deficiency have enzyme levels between 10-30% normal. In the US and many other countries, newborn screening includes tests for BTD deficiency. Approximately 150 mutations in the BTD gene have been reported to cause biotinidase deficiency. The BTD gene is located on chromosome 3p25. A mouse model of biotinidase deficiency has been developed to study various aspects of the disorder. [23, 24]
Holocarboxylase Synthetase (HCLS) Deficiency is also an autosomal recessive disorder and can be diagnosed prenatally. As discussed earlier, the enzyme HCLS is required for the biotinylation of the apocarboxylase enzymes into the active holocarboxylase forms; therefore deficiency leads to multiple carboxylase deficiency. Infants with this disorder present in the first few months of life with acidosis, hyperammonemia, hypotonia, seizures and developmental delay. Mutations in the HCLS gene cause HCLS deficiency.
Because both biotinidase and HCLS deficiency leads to decreased levels of the biotin-dependent carboxylases, the two conditions have also been classified as multiple carboxylase deficiency. Profound biotinidase deficiency was previously known as early-onset multiple carboxylase deficiency, partial biotinidase deficiency as late-onset or juvenile-onset multiple carboxylase deficiency and HCLS deficiency as neonatal or early-onset multiple carboxylase deficiency. [25, 26, 27]
Rarely, isolated deficiencies of each of the five individual biotin-dependent carboxylases may also occur.
Biotin deficiency due to a defect in biotin transport has also been described. [28]
Regardless of the etiology of biotin deficiency, clinical manifestations are similar. However, the age of onset, rates of symptom development and the sequence in which symptoms appear can greatly differ. All of the mechanisms responsible for the development of the manifestations have not been established.
Epidemiology
Incidence
As mentioned previously, dietary deficiency of biotin is uncommon.
Worldwide the incidence of profound biotinidase deficiency is estimated to be 1 in 137,401; the incidence of partial biotinidase deficiency, 1 in 109,921 and the overall incidence, 1 in 61,067. [27] In populations with high rates of consanguinity (e.g., Saudi Arabia, Turkey) the incidence is higher. Neto et al noted that the estimated incidence of biotinidase deficiency in Brazil is about 1 case per 9,000 population. [29] In the US the incidence is reported to be higher in Hispanics and lower in African Americans. In the general population, 1 in 120 is a carrier.
The incidence of holocarboxylase synthetase deficiency is estimated to be 1 in 87,000.
Race-, sex-, and age-related demographics
Biotin deficiency can occur in individuals of any race; however, as mentioned above, genetic disorders affecting biotin metabolism may affect certain races and ethnic groups disproportionately.
Biotin deficiency occurs with equal frequency in both sexes.
Signs and symptoms of biotin deficiency can develop in persons of any age.
Prognosis
The prognosis in most cases is excellent when biotin deficiency is promptly detected and treated.
Once biotin therapy has been initiated with the proper dosage, most signs and symptoms of biotin deficiency should begin to disappear within 3-5 weeks and completely resolve within 2-3 months.
Children who have developed sensorineural hearing loss secondary to profound biotinidase deficiency usually do not have improved hearing after biotin treatment. Also, optic atrophy and developmental delay are usually irreversible.
Holocarboxylase synthetase deficiency is biotin-responsive; however, the isolated carboxylase deficiencies are not.
With early detection and biotin therapy, many of the symptoms and signs of biotin deficiency are reversible. However, if left untreated, vision problems, hearing loss, and developmental delay can occur and these are usually irreversible. [30] Dietary biotin deficiency, biotinidase deficiency, and holocarboxylase synthetase deficiency all respond to biotin treatment; however isolated carboxylase deficiencies are not biotin-responsive. [27]
Complications
Irreversible complications that may develop if treatment is delayed or inadequate include vision problems, sensory-neural hearing loss, ataxia, cognitive impairment, and developmental delay. [31, 32, 33, 30]
Fungal infections are common at initial presentation and need to be treated with appropriate antifungal medications. In those where the diagnosis has been delayed and irreversible complications have occurred, deficits may persist despite biotin therapy. Complications that may be irreversible include vision and hearing loss, and developmental and cognitive delay.
Patient Education
Patients at risk of dietary deficiency of biotin (chronic alcoholics, those who are pregnant or lactating, those consuming excessive egg whites, those on chronic antibiotics or anticonvulsants) should be appropriately counseled and biotin supplements offered if appropriate.
Genetic counseling should be offered when the cause is a genetic disorder, such as biotinidase deficiency or holocarboxylase synthetase deficiency, both of which are inherited in an autosomal recessive manner. Genetic testing of asymptomatic siblings should be offered to ensure early detection and treatment. [34] At-risk relatives may also be tested to determine carrier status. Prenatal testing and preimplantation genetic diagnosis may be offered if appropriate.
Intake of biotin supplements may lead to interference with certain laboratory tests; it is advisable for patients to inform their health care providers if they are taking any supplements that contain biotin. [15]
Instruct patients regarding the dangers of consuming raw eggs.
Instruct patients who are receiving certain anticonvulsant medications regarding signs and symptoms of biotin deficiency so that they can seek medical attention should signs and symptoms develop.
-
Biotin is a bicyclic (more precisely, heterocyclic) compound composed of an ureido ring (A) fused with a tetrahydrothiophene ring (B). A valeric acid substituent is attached to one of the carbon atoms of the tetrahydrothiophene ring.
-
Carboxybiotin carboxylase is the activated form of a carboxylase that conducts the actual carboxylation of a substrate. The CO2 residue attached to the nitrogen atom diagonally across from the valeric acid substituent is transferred to the substrate to be carboxylated, and the original carboxylase is liberated intact.
-
Depiction of the flow of biotin in the biotin cycle.
-
Biocytin is the product of the complete proteolysis of biotin-containing proteins and peptides. The enzyme biotinidase cleaves biocytin into free biotin and the amino acid lysine. The free biotin is then available for intestinal absorption or intracellular coupling to an apocarboxylase to form a holocarboxylase.
-
The biotin molecule is bound to the protein by a peptide bond to an e-amino group of an apocarboxylase to form a holocarboxylase.





