Background
Cholestasis is defined as a decrease in bile flow due to impaired secretion by hepatocytes or to obstruction of bile flow through intra-or extrahepatic bile ducts. Therefore, the clinical definition of cholestasis is any condition in which substances normally excreted into bile are retained. Serum concentrations of conjugated bilirubin, bile salts, and gamma glutamyl transferase are the labs most commonly measured to assess for cholestasis.
Not all substances normally excreted into bile are retained to the same extent in various cholestatic disorders. In some conditions, serum bile salts may be markedly elevated while bilirubin is only modestly elevated and vice versa. However, demonstrable retention of several substances is needed to establish a diagnosis of cholestasis. Only in rare disorders of bilirubin metabolism (eg, Dubin-Johnson syndrome, Rotor syndrome) does an isolated increase in the serum concentration of conjugated bilirubin appear, so increased serum conjugated bilirubin indicates cholestasis. The histopathologic definition of cholestasis is the appearance of bile within the elements of the liver, usually associated with secondary cell injury.
Pathophysiology
The mechanisms of cholestasis can be broadly classified into hepatocellular, where an impairment of bile formation occurs, and obstructive, where impedance to bile flow occurs after it is formed. The typical histopathologic features of hepatocellular cholestasis include the presence of bile within hepatocytes and canalicular spaces, in association with generalized cholate injury. Typical of obstructive cholestasis is bile plugging of the interlobular bile ducts, portal expansion, and bile duct proliferation in association with centrilobular cholate injury. [1]
Bile is a highly complex water-based medium containing inorganic ions and many classes of organic amphiphiles. The formation of bile involves multiple mechanisms and levels of regulation. The transport of solute into the canaliculus by specific transporters creates chemical and osmotic gradients and promotes water flow by a paracellular pathway. Several of these specific transporters have been identified, and their function has been characterized. The identification of defective transporters in some familial cholestatic disorders has led to improved understanding of the molecular mechanisms of human cholestasis as well as potential therapeutic agents. [2, 3, 4]
Redundancies in the mechanisms of solute transport result in bile formation and the absence or impairment of a single transporter is not expected to result in the failure of bile formation. Instead, a process of amplification is required to produce clinical cholestasis. A primary mechanism of amplification is the retention of hydrophobic bile salts, strong detergents that cause membrane injury and impairment of membrane function. Retained bile salts down-regulate new bile acid synthesis, which results in a reduction of the bile salt pool and in reduced enterohepatic recirculation.
Retention of cholesterol results in increased cholesterol content of membranes that reduces their fluidity and impairs the function of integral membrane proteins. These amplification mechanisms result in further retention of damaging substances, accelerated membrane injury and dysfunction, and ultimately, generalized failure of the excretory mechanism for bile. This converging pathway makes the differentiation of cholestatic diseases on clinical grounds very difficult.
Obstructive cholestasis is usually the result of physical obstruction of the biliary system at the level of the extrahepatic bile ducts. However, obstruction or paucity of small bile ducts can result in functional obstruction of the entire biliary system. This is the mechanism involved in the cholestasis observed in Alagille syndrome, which is associated with heart, skeleton, eye, kidney, and facial manifestations. The morbidity and prognosis of Alagille syndrome mainly depends on the severity of the patient's liver and heart diseases.
Retention of bile salts results in injury to biological membranes throughout the body; the liver is the most affected. The retention of hydrophobic bile salts results in their incorporation into membranes, which alters membrane fluidity and function. Bile salt injury of hepatocyte membranes is an important amplifier of cholestasis. The retention of secondary cholestatic bile acids, such as lithocholic acid, results in further membrane injury.
The differential diagnosis of cholestasis in neonates and infants is much broader than in older children and adults. In the neonatal period, the immature liver is relatively sensitive to injury, and the response of the immature liver is more limited owing to an immaturity of hepatobiliary function. Cholestasis develops in response to a wide variety of insults. In the neonatal period, cholestasis is considered the result of immaturity of several critical mechanisms of bile formation. So-called physiologic cholestasis of infancy results from immaturity of these mechanisms. Early initiation of enteral feedings as tolerated is the best way to prevent and manage cholestasis in preterm infants. [5]
The effects of cholestasis are profound and widespread. Although the principal effects involve the function of the liver and intestine, secondary effects can involve every organ system. The primary effects are bile retention, regurgitation of bile into serum, and reduction in bile delivery to the intestine. These result in secondary effects that lead to worsening liver disease and systemic illness. Pruritus secondary to cholestasis is a significant cause of morbidity for patients and is challenging to treat.
Retention of conjugated bilirubin and its regurgitation into serum
Excretion of conjugated bilirubin is the rate-limiting step of bilirubin clearance. During cholestasis, conjugation of bilirubin continues but excretion is reduced. The mechanism by which conjugated bilirubin regurgitates into serum is unclear, but it may differ according to the disease etiology. In hepatocellular cholestasis, where bile formation is reduced, conjugated bilirubin is likely to efflux directly from the hepatocyte via diffusion or vesicular exocytosis. On the other hand, in obstructive cholestasis, conjugated bilirubin possibly enters the canalicular space and effluxes back through a weakened tight junction.
The presence of elevated serum concentration of conjugated bilirubin is a principal sign of cholestasis. It results in jaundice, which can be detected by scleral icterus at a level as low as 2 mg/dL, and by dark urine. The level of conjugated bilirubin is affected by the rate of production of bilirubin, the degree of cholestasis, and alternate pathways of elimination, principally renal excretion.
Alternate elimination pathways, principally by way of the kidneys, limit the absolute elevation of conjugated bilirubin. Conjugated bilirubin levels rarely exceed 30 mg/dL, although such elevated levels can occur. Because conjugated bilirubin is relatively weakly bound to albumin, it can dissociate relatively easily and be filtered into the urine. Patients with cholestasis may report dark urine and infants with cholestasis may have a stained diaper, thus making examination of the urine a useful starting point in the evaluation of an infant with jaundice.
Increased serum concentration of nonconjugated bilirubin
Increased serum concentration of unconjugated bilirubin is present in most patients with cholestasis. The rate of bilirubin conjugation is likely reduced by end-product inhibition or as the result of hepatocyte injury. The rate of bilirubin production may also be increased as the result of hemolysis that can accompany cholestasis.
Newer methods of measuring bilirubin in serum have resulted in the discovery of a fraction of serum bilirubin that is covalently bound to albumin, known as delta fraction of bilirubin or biliprotein. This fraction may account for a large proportion of total bilirubin in patients with cholestatic jaundice but is absent in patients with nonconjugated hyperbilirubinemia.
Patients are considered to have conjugated hyperbilirubinemia when their conjugated bilirubin level is >1mg/dL if the total bilirubin level is >5mg/dL. When the total bilirubin level is < 5mg/dL, conjugated hyperbilirubinemia is defined as a conjugated bilirubin level of >20% of the total bilirubin level.
Hypercholemia
Hypercholemia, or increased serum bile salt concentration, is a universal consequence of cholestasis. [6] The transport of bile salts from plasma to bile is the principal driving force for bile formation. Failure to transport bile salts may be a principal mechanism of cholestasis or may be a consequence of the effects of cholestasis on hepatocyte function. In either case, the liver cell retains bile salts, resulting in down-regulation of new bile acid synthesis and in an overall reduction in the total pool size. Bile salts are regurgitated from the hepatocyte, which results in an increase in the concentration of bile salts in the peripheral circulation. Furthermore, the uptake of bile salts entering the liver in portal vein blood is inefficient, which results in spillage of bile salts into the peripheral circulation.
Overall, patients with cholestasis have an increase in serum concentration of bile salts, an increase in hepatocyte concentration of bile salts, a decrease of bile salts in the enterohepatic circulation, and a decrease in the total bile salt pool size.
Pruritus
A common clinical consequence of cholestasis is pruritus. Several links to its pathogenesis have been proposed, including the role of bile acids, endogenous opioid and serotonins, and lysophosphatidic acid. The mechanism of pruritus in liver disease is not entirely understood, and major debate concerns its relationship to the retention of bile salts. The serum or tissue concentrations of bile salts do not correlate well with the degree of pruritus, although all patients with pruritus related to liver disease have significant elevations of serum bile salts. Therapeutic approaches that reduce pruritus generally also reduce serum bile salt concentrations.
Newer theories suggest that patients have differing sensitivities to elevated bile salt concentrations, which act on peripheral pain afferent nerves to produce the sensation of itching. This stimulation involves opiate-mediated pathways, and opiate antagonists can block cholestasis-associated itching. Itching does not appear to be associated with histamine release, and antihistamine therapy is generally ineffective. Ultraviolet B phototherapy has been successfully used to treat pruritus.
Decock et al have reported that ultraviolet B phototherapy appears to be a promising and well-tolerated treatment for cholestasis-associated pruritus. [7] Newer treatment options include ileal bile acid transporter inhibitors (IBATi), which function by inhibiting the reuptake of bile acids in the terminal ileum. Two drugs are currently FDA-approved: odevixibat for progressive familial intrahepatic cholestasis and maralixibat for Alagille syndrome. Additional IBATi drugs are in development, including linerixibat for primary biliary cholangitis and volixibat for intrahepatic cholestasis of pregnancy. Common side effects include dose-dependent diarrhea and abdominal discomfort. [8]
Patients with cholestasis experience a range of severity of pruritus; some people experience serious impairments in their quality of life. Scratching is the most measurable effect of pruritus. The degree of pruritus can be quantitated by clinical findings related to scratching, which has been useful in monitoring patient response to therapy.
Hyperlipidemia
Hyperlipidemia is characteristic of some but not all cholestatic diseases. Serum cholesterol is elevated in cholestasis because its metabolic degradation and excretion are impaired. Bile is the normal excretory pathway for cholesterol, and with reduced bile formation, cholesterol is retained. Cholesterol retention can cause an increase in membrane cholesterol content and a reduction in membrane fluidity and membrane function, thereby amplifying the cholestasis. Furthermore, bile salts are the metabolic products of cholesterol, and in cholestasis, synthesis of bile salts is reduced.
The elevated cholesterol associated with cholestasis is due to a deficiency in the lecithin-acyltransferase enzyme that leads to a formation of lipoprotein X. Lipoprotein X contains little to no apoB and thus, theoretically, should not cause arteriosclerosis. Lipoprotein X is metabolized via a unique pathway as it is not taken up by LDL receptors, thus preventing feedback inhibition of cholesterol synthesis and perhaps accounting for elevated cholesterol levels observed in patients with cholestasis. As such, use of oral bile salt–binding agents, such as cholestyramine, has little effect on serum cholesterol in this setting. Agents that block the synthesis of cholesterol have been used sparingly in cholestasis and cannot be recommended at this time. The proper approach to treating hypercholesterolemia in cholestatic liver disease is to treat the liver disease itself. [9, 10]
The contribution of dietary cholesterol to the elevated serum cholesterol in patients with cholestasis is probably minimal, and limiting the diet in order to reduce serum cholesterol is not justified because that maneuver may have secondary effects on nutrition.
Xanthomas
Xanthomas may result from the deposition of cholesterol into the dermis. The development of xanthomas is more characteristic of obstructive cholestasis than of hepatocellular cholestasis. Xanthomas may develop rapidly over a few months in acute extrahepatic biliary obstruction. Acutely developing xanthomas are usually the eruptive type, which are white pustular lesions pinpoint to 2 mm in diameter, that appear first on the trunk and in the diaper area. See the image below.
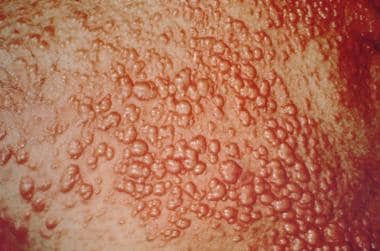 Cholestasis. Eruptive xanthomas. Courtesy of Duke University Medical Center.
Cholestasis. Eruptive xanthomas. Courtesy of Duke University Medical Center.
Failure to thrive
One of the major clinical effects of cholestasis, particularly chronic cholestasis, is failure to thrive. The mechanisms of failure to thrive include malabsorption, anorexia, poor nutrient use, hormonal disturbances, and secondary tissue injury. Malabsorption in cholestatic liver disease results from reduced delivery of bile salts to the intestine, which results in inefficient digestion and absorption of fats. Digestion is affected because bile salts are important for the function of bile salt–dependent lipase activity and the stabilization of the lipase-colipase complex. In addition, bile salts are important in stabilization of lipid emulsions, which is important for increasing the surface area on which lipase works.
Absorption is inefficient because of reduced formation of intestinal micelles, which are important for removing the end products of lipolysis and effecting their absorption. The result of these events is the malabsorption of fat and fat-soluble vitamins.
Malabsorption of fat results in the loss of a source of calories that is important in infant nutrition. Furthermore, the delivery of fat into the colon can result in colonic secretion and diarrhea. Adults with fat malabsorption often experience anorexia. This may also occur in infants, but more often, infants take increased amounts of formula to compensate for loss of calories. Finally, the loss of fat into the stool also results in calcium wasting through the formation of calcium soaps of fatty acids. This may play an important role in bone disease in children and adults with chronic cholestasis.
The treatment of fat malabsorption principally involves dietary substitution. In older patients, a diet that is rich in carbohydrates and proteins can be substituted for a diet containing long-chain triglycerides. In infants, substitution may not be possible, and the substitution of a formula containing medium-chain triglycerides may improve fat absorption and nutrition.
The malabsorption of fat-soluble vitamins can result in vitamin deficiency states. Vitamins E, D, K, and A are all malabsorbed in cholestasis, and in that order. [11] Vitamin E deficiency can result in peripheral neuropathy and possibly hemolysis. Vitamin D deficiency results in osteomalacia and rickets. Vitamin K deficiency causes coagulopathy and possibly reduced brain development. Vitamin A deficiency does not result in clinical disease in cholestasis. In chronic cholestasis, careful attention must be paid to prevent fat-soluble vitamin deficiencies. This is accomplished by administering fat-soluble vitamins and monitoring the response to therapy. [12]
Etiology
Causes of cholestasis include the following:
-
Obstructive cholestasis
Biliary atresia
Congenital bile duct anomalies (choledochal cysts)
Infectious cholangitis (cholangitis)
Cholangitis associated with Langerhans cell histiocytosis
Alagille syndrome
Nonsyndromic ductal paucity
-
Hepatocellular cholestasis
Hepatitis (hepatitis A, hepatitis B, hepatitis C)
Inborn errors of bile acid synthesis
Drug-induced cholestasis
Total parenteral nutrition (TPN)–associated cholestasis
Progressive familial intrahepatic cholestasis [13, 14, 15, 16, 17]
Epidemiology
Sex- and age-related demographics
No clear difference in the incidence of cholestasis between males and females is observed. Incidence is equal in most genetic diseases leading to cholestasis. However, several conditions have a female dominance, including biliary atresia, [18] drug-induced cholestasis, and of course, cholestasis of pregnancy.
Cholestasis is observed in people of every age group. However, newborns and infants are more susceptible and more likely to develop cholestasis as a consequence of immaturity of the liver.
Young gestational age, low birth body weight, more sepsis episodes, and long duration of parenteral nutrition are risk factors associated with parenteral nutrition-associated cholestasis. [19]
Prognosis
Prognosis for patients is dependent on the cause of cholestasis, with patients with biliary atresia being among those having the greatest risk of progressing to liver cirrhosis and requiring a liver transplant.
Cholestasis is not a primary cause of death. However, it is the cause of considerable morbidity as indicated in Pathophysiology.
-
Cholestasis. Eruptive xanthomas. Courtesy of Duke University Medical Center.