Practice Essentials
Congenital diaphragmatic hernia (see the image below) is characterized by a variable degree of pulmonary hypoplasia associated with a decrease in cross-sectional area of the pulmonary vasculature and alterations of the surfactant system. There are three basic types of congenital diaphragmatic hernia: the posterolateral Bochdalek hernia (occurring at approximately 6 weeks' gestation), the anterior Morgagni hernia, and the hiatus hernia.
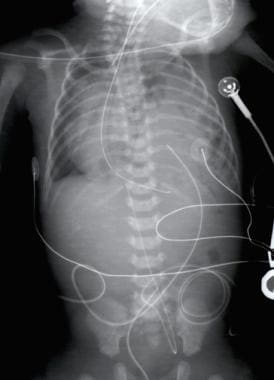 Pediatric Congenital Diaphragmatic Hernia. Radiograph of a 1-day-old infant with a moderate-sized congenital diaphragmatic hernia (CDH). Note the air- and fluid-filled bowel loops in the left chest, the moderate shift of the mediastinum into the right chest, and the position of the orogastric tube.
Pediatric Congenital Diaphragmatic Hernia. Radiograph of a 1-day-old infant with a moderate-sized congenital diaphragmatic hernia (CDH). Note the air- and fluid-filled bowel loops in the left chest, the moderate shift of the mediastinum into the right chest, and the position of the orogastric tube.
Signs and symptoms
Infants with congenital diaphragmatic hernias most commonly present with respiratory distress and cyanosis in the first few minutes or hours of life, although a later presentation is possible. The respiratory distress can be severe and may be associated with circulatory insufficiency, requiring aggressive resuscitative measures.
See Clinical Presentation for more detail.
Diagnosis
Examination in infants with congenital diaphragmatic hernias include the following findings:
-
Scaphoid abdomen
-
Barrel-shaped chest
-
Respiratory distress (retractions, cyanosis, grunting respirations)
-
In left-sided posterolateral hernia: Poor air entry on the left, with a shift of cardiac sounds over the right chest; in patients with severe defects, signs of pneumothorax (poor air entry, poor perfusion) may also be found
-
Associated anomalies: Dysmorphisms such as craniofacial abnormalities, extremity abnormalities, or spinal dysraphism may suggest syndromic congenital diaphragmatic hernia
Laboratory tests
Laboratory studies that may be indicated in congenital diaphragmatic hernia include the following:
-
Arterial blood gas (ABG) measurements: To assess for pH, PaCO2, and PaO2
-
Serum lactate: May be helpful for assessing for circulatory insufficiency or severe hypoxemia associated with tissue hypoxia
-
Chromosome studies, including microarray analysis
-
levels of serum electrolytes, ionized calcium, and glucose
Continuous pulse oximetry is valuable in the diagnosis and management of persistent pulmonary hypertension of the newborn.
Imaging studies
The following radiologic studies may be used to evaluate congenital diaphragmatic hernia:
-
Chest radiography: To confirm diagnosis of congenital diaphragmatic hernia and to rule out pneumothorax
-
Cardiac ultrasonography: To rule out cardiac anomalies
-
Echocardiography: To assess myocardial function and determine whether left ventricular mass is significantly decreased
-
Renal ultrasonography: To rule out genitourinary anomalies
-
Cranial magnetic resonance imaging: When considering extracorporeal support to evaluate for intraventricular bleeding and hypoxic-ischemic changes, as well as to rule out major intracranial anomalies
-
Cranial sonography: When an infant is considered for extracorporeal support
Procedures
-
Endotracheal intubation and mechanical ventilation: Required in all infants with severe congenital diaphragmatic hernia who present in the first hours of life
-
Placement of an indwelling catheter in the umbilical artery or in a peripheral artery (radial, posterior tibial): For continuous blood pressure and frequent ABG monitoring
-
Placement of a venous catheter via the umbilical vein: To allow for administration of inotropic agents and hypertonic solutions (eg, calcium gluconate)
-
Venoarterial or venovenous extracorporeal membrane oxygenation (ECMO) support
-
Biopsy may be needed for rare chromosomal disorders that can be diagnosed only based on skin biopsy findings
See Workup for more detail.
Management
Ideally, fetuses carrying a diagnosis of congenital diaphragmatic hernia should be delivered at an ECMO center. Sometimes stabilization for transfer can be very challenging. Medical therapy in patients with congenital diaphragmatic hernia is directed toward optimizing oxygenation while avoiding barotrauma. [1] Management includes the following:
-
Placement of a vented orogastric tube and connecting it to continuous suction to prevent bowel distention and further lung compression
-
Avoiding mask ventilation and immediately intubating the trachea
-
Avoiding high peak inspiratory pressures with mechanical ventilation; synchronizing ventilation with the infant's respiratory effort
-
Continuous monitoring of oxygenation, blood pressure, and perfusion
-
Maintaining glucose and ionized calcium concentrations within reference range
Surgery
Fetal surgical intervention (fetal repair, fetal tracheal occlusion) for congenital diaphragmatic hernia may not improve survival compared with standard therapy. [2, 3] Postnatal procedures include the following:
-
Reduction of the herniated viscera and closure of the diaphragmatic defect
-
Chest tube drainage in the presence of a tension pneumothorax
-
Transplantation of a single lung (single case report)
The ideal time to repair a congenital diaphragmatic hernia is unknown. Some authors suggest that repair 24 hours after stabilization is ideal, but delays of up to 7-10 days are typically well tolerated, and many surgeons now adopt this approach. Other surgeons prefer to operate on these neonates when normal pulmonary artery pressure is maintained for at least 24-48 hours based on echocardiography.
Pharmacotherapy
The following medications may be used to help stabilize blood pressure and circulating volume, alleviate pulmonary distress, and/or correct hypoxemia in infants with congenital diaphragmatic hernia:
-
Vasoactive agents (eg, dopamine, dobutamine, milrinone)
-
Opioid analgesics (eg, fentanyl)
-
Neuromuscular relaxing agents (eg, pancuronium, vecuronium)
-
Pulmonary vasodilating agents (eg, nitric oxide)
See Treatment and Medication for more detail.
Background
The topic of congenital diaphragmatic hernia (CDH) has frequently appeared in the medical literature since its first description in the early 18th century. Initial theories about the pathophysiology of this condition centered on the presence of the herniated viscera within the chest and the need for its prompt removal.
In 1946, Gross reported the first successful repair of a neonatal diaphragmatic hernia in the first 24 hours of life. [4] The medical literature for the next decade addressed congenital diaphragmatic hernia as a surgical problem and discussed various technical aspects of surgical repair, including techniques required to close large defects. In the 1960s, however, Areechon and Reid observed that the high mortality rate of congenital diaphragmatic hernia was related to the degree of pulmonary hypoplasia at birth. [5]
Over the past 20 years, pulmonary hypertension and pulmonary hypoplasia have been recognized as the 2 cornerstones of the pathophysiology of congenital diaphragmatic hernia. In recent years, evidence suggests that cardiac maldevelopment may further complicate the pathophysiology of congenital diaphragmatic hernia. [6] See the image below.
Pediatric Congenital Diaphragmatic Hernia. Radiograph of a 1-day-old infant with a moderate-sized congenital diaphragmatic hernia (CDH). Note the air- and fluid-filled bowel loops in the left chest, the moderate shift of the mediastinum into the right chest, and the position of the orogastric tube.
Pathophysiology
The 3 basic types of congenital diaphragmatic hernia include the posterolateral Bochdalek hernia (occurring at approximately 6 weeks' gestation), the anterior Morgagni hernia, and the hiatus hernia. The left-sided Bochdalek hernia occurs in approximately 85% of cases. Left-sided hernias allow herniation of both the small and large bowel and intraabdominal solid organs into the thoracic cavity. In right-sided hernias (13% of cases), only the liver and a portion of the large bowel tend to herniate. Bilateral hernias are uncommon and are usually, but not always, fatal. [7, 8]
Congenital diaphragmatic hernia is characterized by a variable degree of pulmonary hypoplasia associated with a decrease in cross-sectional area of the pulmonary vasculature and alterations of the surfactant system. The lungs have a small alveolar capillary membrane for gas exchange, which may be further decreased by surfactant dysfunction. In addition to parenchymal disease, increased muscularization of the intraacinar pulmonary arteries appears to occur. In very severe cases, left ventricular hypoplasia is observed. Pulmonary capillary blood flow is decreased because of the small cross-sectional area of the pulmonary vascular bed, and flow may be further decreased by abnormal pulmonary vasoconstriction.
Etiology
The diaphragm initially develops as a septum between the heart and liver, progresses posterolaterally, and closes at the left Bochdalek foramen at approximately 8-10 weeks' gestation. [9]
The herniation of viscera in severe congenital diaphragmatic hernia is believed to occur during the pseudoglandular stage of lung development. Lung compression results in pulmonary hypoplasia that is most severe on the ipsilateral side, although both lungs may be abnormal. Pulmonary hypoplasia is associated with fewer bronchial generations, alveoli, and arterial generations.
Congenital diaphragmatic hernia can be induced in rat models with administration of the herbicide toxin nitrofen. Studies in these models show that the diaphragmatic defect occurs in the initial stages of diaphragm development, rather than in the later stages.
Fetal exposure to nitrofen causes a variable amount of lung hypoplasia. The fact that only 60-90% of exposed rat pups demonstrate diaphragmatic defects suggests a “dual-hit” hypothesis, in which 2 insults (one primarily affecting the lungs and another primarily affecting diaphragm development) contribute to the pathophysiology of congenital diaphragmatic hernia.
Congenital diaphragmatic hernia may occur as a nonsyndromic or isolated defect. Less than 2% of such cases are estimated to be familial. Pedigrees consistent with autosomal recessive, autosomal dominant, and X-linked inheritance patterns have been described.
More than 10% of infants with congenital diaphragmatic hernia have an underlying syndromic diagnosis, although few gene mutations are currently recognized. Congenital diaphragmatic hernia is a recognized finding of Cornelia de Lange syndrome, an autosomal dominant syndrome with characteristic facial features, hirsutism, and developmental delay. Fryns syndrome is an autosomal recessive condition that includes congenital diaphragmatic hernia as the cardinal feature, along with hypoplasia of the distal digits and other variable abnormalities of the brain, heart, and genitourinary development. An associated gene has not yet been identified, and the prognosis of Fryns syndrome is poor.
Chromosome abnormalities have been reported in as many as 30% of infants with congenital diaphragmatic hernia, which has been described as part of trisomy 13, trisomy 18, trisomy 21, and Turner syndrome (monosomy X). Pallister-Killian syndrome (tetrasomy 12p mosaicism) presents with findings that are similar to those of Fryns syndrome, including coarse facial features, aortic stenosis, cardiac septal defects, and abnormal genitalia. This diagnosis can only be made if a karyotype is determined based on skin biopsy findings.
Chromosome deletions on chromosomes 1q, 8p, and 15q have been reported in association with congenital diaphragmatic hernia. Deletions of chromosomes 8p and 15q appear to be associated with heart malformations.
Deficiencies in vitamin A availability, metabolism, and signaling have been found to contribute to the development of congenital diaphragmatic hernia in animal models and may also be relevant in human fetal development. [10]
Epidemiology
International data
Congenital diaphragmatic hernia occurs in 1 of every 2000-3000 live births and accounts for 8% of all major congenital anomalies. The risk of recurrence of isolated (ie, nonsyndromic) congenital diaphragmatic hernia in future siblings is approximately 2%. [11] Familial congenital diaphragmatic hernia is rare (< 2% of all cases), and both autosomal recessive and autosomal dominant patterns of inheritance have been reported. Congenital diaphragmatic hernia is a recognized finding in Cornelia de Lange syndrome and also occurs as a prominent feature of Fryns syndrome, an autosomal re
Sex- and age-related demographics
Most studies report that congenital diaphragmatic hernia occurs equally in males and females.
Although congenital diaphragmatic hernia is usually a disorder of the newborn period, as many as 10% of patients may present after the newborn period and even during adulthood. A 2020 retrospective analysis of 2015-2018 data from the American College of Surgeons National Surgical Quality Improvement Program (ACS NSQIP) database found 110 adult patients with congenital diaphragmatic hernia who underwent surgical correction. [12]
Outcome in patients with late presentation of congenital diaphragmatic hernia is extremely good, with low or no mortality. cessive disorder with variable features, including diaphragmatic hernia, cleft lip or palate, and distal digital hypoplasia.
Prognosis
Overall reported survival varies among institutions. Keep in mind that a single institution's results may look better than those provided by population-based studies because of case-selection biases. When all resources, including extracorporeal membrane oxygenation (ECMO), are provided, reported survival rates range from 40% to 90%. The Extracorporeal Life Support Organization (ELSO) registry reports the ECMO overall survival to hospital discharge at 73%; overall survival to decannulation from ECMO support is 83%. [13] Complications of neonatal ECMO include hemolysis, hemorrhage, and clotting. [13]
As noted, survivors are at risk for significant long-term morbidity, including chronic lung disease, growth failure, gastroesophageal reflux, hearing loss, and neurodevelopmental delay. The risk appears to be highest in infants with severe lung disease (need for long term supplemental oxygen), need for patch closure of the diaphragm, and need for gastrostomy tube feeding.
Morbidity/mortality
Mortality has traditionally been difficult to determine. This is partially because of the "hidden mortality" for this condition, which refers to infants with congenital diaphragmatic hernia who die in utero or shortly after birth, prior to transfer to a surgical site. This bias may be especially important when evaluating institutional reports of outcome.
A population-based study from Western Australia indicated that only 61% of infants with congenital diaphragmatic hernia are live born. In that study, nearly 33% of pregnancies that involved a fetus with congenital diaphragmatic hernia were electively terminated. Most of the pregnancies (71%) were terminated because of the presence of another major anomaly.
Mortality after live birth is generally reported to range from 40% to 62%, and some authors argue that the true mortality of congenital diaphragmatic hernia has not changed with introduction of new therapies. The presence of associated anomalies has consistently been associated with decreased survival; other associations with poor outcome include prenatal diagnosis, prematurity, low birth weight, and early pneumothorax.
Keller et al found that infants with congenital diaphragmatic hernia who have poor outcomes (death or discharge on oxygen) have higher plasma levels of endothelin-1, which is dysregulated in pulmonary hypertension. [14] Severity of pulmonary hypertension was also associated with increasing endothelin-1 levels.
In a 2020 retrospective analysis of 2015-2018 data from the American College of Surgeons National Surgical Quality Improvement Program (ACS NSQIP) database, of 110 adult patients with congenital diaphragmatic hernia who underwent surgical correction, 4.55% required reintervention, 5.45% had postoperative respiratory failure, and 3.64% required reintubation, with no deaths. [12] Although there was no difference between those that underwent open repair and those who had minimally invasive repair with regard to procedure length, discharge to home, or postoperative complications, the investigators noted a longer length of stay in patients in the open repair group relative to the minimally invasive repair group, as well as more use of mesh in the minimally invasive group than in the open repair group. [12]
-
Pediatric Congenital Diaphragmatic Hernia. Radiograph of a 1-day-old infant with a moderate-sized congenital diaphragmatic hernia (CDH). Note the air- and fluid-filled bowel loops in the left chest, the moderate shift of the mediastinum into the right chest, and the position of the orogastric tube.







