Background
The clinical features of neurofibromatosis type 1 (NF1; also referred to as peripheral neurofibromatosis), [1] the most common form of the disease, were reported in several family members by the German pathologist Virchow in 1847, but it was his student von Recklinghausen who 35 years later described the histologic features of the syndrome that often bears his eponym (ie, von Recklinghausen disease).
The orthopedic manifestations and, especially, the complications after treatment of NF1 are common and have a prominent place in the orthopedic literature. The intent of this article is to identify the complications most commonly associated with the orthopedic manifestations of neurofibromatosis and to present strategies for their management. [2, 3, 4]
Skeletal complications of NF1 can be categorized as generalized or focal manifestations.
Generalized skeletal abnormalities include osteoporosis/osteopenia, osteomalacia, shortness of stature, and macrocephaly. The focal orthopedic complications of neurofibromatosis, which usually appear early, include spinal deformities such as scoliosis and kyphoscoliosis, congenital bowing and pseudoarthrosis of the tibia and the forearm, chest-wall deformities, overgrowth phenomenon of the extremity, and soft-tissue tumors. (See the images below.) Focal abnormalities of the skeleton are less common than generalized abnormalities are but may cause significant morbidity. [2, 5]
Neurofibromatosis type 2 (NF2), or central neurofibromatosis, is associated with bilateral vestibular schwannomas and multiple spinal schwannomas. NF1 and NF2 are genetically distinct disorders despite the similarity of their names.
Segmental neurofibromatosis is characterized by features of NF1 involving a single body segment. Typically, only a single segment of the body (such as the left upper extremity) is affected with café-au-lait spots and freckling, and lesions usually do not cross the body midline.
Early neurofibromatosis literature recognized that a mild form of NF1 existed, consisting primarily of familial café-au-lait spots. Since then, multiple families with such mild involvement have been found to have mutations in the SPRED1 gene. This condition, now called Legius syndrome, can present with multiple café-au-lait spots, freckling, macrocephaly, and mild learning disabilities, but does not present with any of the benign or malignant tumors associated with NF1.
Schwannomatosis is a distinct form of neurofibromatosis that typically involves multiple schwannomas throughout the body, but without the vestibular schwannomas typical of NF2. It is most commonly a disease of adulthood that consists of multiple deep painful peripheral nerve sheath tumors, which may occur in a generalized form or in a segmental distribution.
NF2, segmental neurofibromatosis, Legius syndrome, and schwannomatosis do not appear to have any bone involvement or other orthopedic manifestations. As a result, they are not discussed in this article.
Spinal Complications
Spinal complications are the most common orthopedic manifestation of NF1. Between 10% and 33% of children with NF1 have spinal deformity. Deformities include dystrophic and nondystrophic changes. The radiologic appearance of the dystrophic changes includes the following features:
-
Scalloping of the vertebral body margins (3 mm in thoracic vertebrae, 4 mm in lumbar vertebrae, or both)
-
Severe rotation of the apical vertebra
-
Widening of the spinal canal
-
Enlargement of the neural foramina
-
Defective pedicles
-
A paraspinal mass
-
Spindling of the transverse process
-
Rib penciling (ie, the rib being smaller in diameter than the second rib) - The ribs may resemble twisted ribbons
-
Presence of dural ectasia [6]
More than three of these dystrophic features are considered diagnostic of dystrophic scoliosis. In some cases, these changes are the result of intraspinal pathology, such as tumors, [7] meningoceles, or dural ectasia. However, the changes may also occur in persons with entirely normal intraspinal contents. In these persons, primary bone dysplasia accounts for the dystrophic changes. [8]
Spinal changes in individuals with NF1 are usually divided into cervical, thoracic, lumbosacral, and spinal canal pathologies.
Cervical spine changes and associated complications
Features of the cervical spine in patients with NF1 have not received enough attention in the literature. Cervical abnormalities occur much more frequently when a scoliosis or kyphoscoliosis is present in the thoracolumbar region, in which case the examiner's attention is focused on the more obvious deformity.
The manifestations of NF1 can be observed as dystrophic changes in the vertebral body, or they can be due to pathologic alignment. The most common abnormality observed is a severe cervical kyphosis, which in itself is highly suggestive of the disorder.
In a study by Yong-Hing et al, 17 patients with NF1 were found to have cervical abnormalities. [9] Of these, seven were asymptomatic, and the rest had either limited motion or pain in the neck. Four patients had neurologic deficits, which were probably attributed to cervical instability. Four of the 17 patients required fusion of the cervical spine. Curtis described eight patients with paraplegia and NF1. Paraplegia in four of these patients was due to cervical spine instability or intraspinal pathology in the cervical spine. [10]
Attention also should be paid to the C1-2 region. Isu et al. [11] described three patients with NF1 who had C1-2 dislocation with neurologic deficit, and all improved after decompression or fusion. No bony changes in the C1-2 relationship were observed on flexion-extension views in any of these patients. Most of the problems in the cervical spine in this study occurred after excision of the tumors, which included resection of the laminae and posterior elements. Postoperatively, the spine is unstable and tends to develop progressive kyphosis.
All patients with NF1 who undergo surgery, who require endotracheal anesthesia, who undergo halo traction, who complain of neck pain, or who present with neck tumors should undergo a cervical radiographic series. If subluxation is suspected, computed tomography (CT), magnetic resonance imaging (MRI), or both are appropriate. Other reasons for obtaining cervical spinal radiographs in a patient with NF1 include the evaluation of torticollis and dysphagia (symptoms indicating intra- or extraspinal neurofibromas).
The authors have seen significant progressive kyphosis following occipitocervical decompression in young children. Deformity progression is silent and may not be noted. Nothing less than biannual imaging follow-up is mandatory. Any patient with any evidence of instability after an operative procedure on the spine that requires laminectomy should undergo spinal fusion.
Scoliosis
Scoliosis is the most common osseous defect associated with NF1. It may vary in severity from mild and nonprogressive to severe curvatures. The cause of this spinal deformity is unknown, but some have suggested that it is secondary to endocrine disturbances, mesodermal dysplasia, and osteomalacia (a localized neurofibromatous tumor eroding and infiltrating bone).
In a general orthopedic clinic, 2% of patients with scoliosis have neurofibromatosis, whereas in a neurofibromatosis clinic, 10-20% of patients have some disorder of the spine. All preadolescent children with neurofibromatosis should be evaluated with scoliosis screening, or the Adams bend test, to exclude a spinal deformity, which usually occurs earlier in children with neurofibromatosis.
Two primary types of scoliosis are observed in persons with neurofibromatosis: dystrophic and nondystrophic. Dystrophic scoliosis is the short-segmented, focal, sharply angulated type that includes fewer than six spinal segments. It has a tendency to progress to a severe deformity. [12, 13] Dystrophic curves may be associated with kyphosis and have a higher incidence of neurologic injury.
The second type of curvature, nondystrophic scoliosis, is similar to the idiopathic curvature observed in adolescents. This form usually involves eight to 10 spinal segments. The deformity is most often convex to the right; however, this is not consistent. Compared to dystrophic curves, nondystrophic curves tend to present in older children with less angulation and rotation of the deformity.
Patients more likely to develop progressive scoliosis are children younger than 7 years who have thoracic lordosis (sagittal plane angle < 20°) and paravertebral tumors. Curves that acquire either three or more penciled ribs or a combination of any three dystrophic features as described above (modulation theory), will almost certainly progress. Durrani et al [14] defined modulation as a process by which a nondystrophic curve acquires the features of a dystrophic curve and behaves as a dystrophic curve. They reported that modulation occurred in about 65% of their patients.
Subsequent MRI studies questioned the theory of modulation. Patients with radiographically labeled nondystrophic curves were found to have significant dysplastic changes on MRI. Given that MRI has higher sensitivity for identifying dystrophic features than x-rays do, it is strongly recommended to characterize the curve as dystrophic or not on the basis of a combination of MRI and x-ray findings. [15]
Meningoceles, pseudomeningoceles, dural ectasia, and dumbbell lesions are all related to the presence of neurofibroma or abnormal pressure phenomena in and around the spinal canal neuraxis. High-volume or contrast MRI should be used in the investigation of all dystrophic curves prior to treatment. Occasionally, these intraspinal elements may directly compromise the cord when instrumentation and stabilization are attempted, or they may cause erosive changes in the bone, preventing primary fusion.
The cervical spine should be evaluated with the initial scoliosis investigation. Evidence of dystrophic changes may be present on a true lateral view. Progressive cervical kyphosis is usually apparent after excision of the posterior elements. The patient presents with a neck deformity after anterior and posterior excision of a neck mass. If any suspicious area is noted on plain radiographs, right and left oblique views should be obtained to look for widening of the neuroforamina. These may represent dumbbell lesions (ie, widening of the neuroforamina caused by dural ectasia or the exit of a neurofibroma from the spinal canal).
Kyphosis
Kyphosis observed in individuals with NF1 is distinguished by acute anteroposterior angulation. Vertebral bodies may be deformed so severely that they are confused with congenital deformities. It may occur by gradual scoliotic rotation and progression or it can be found early in the disease with an abrupt angular kyphotic curve. Severe kyphosis is the most common cause of neurologic deficits in NF1.
Lordoscoliosis
Lordoscoliosis has not been as frequently reported in patients with NF1 as kyphoscoliosis has. However, lordosis of the thoracic spine predisposes to significant respiratory compromise and mitral valve prolapse.
Spondylolisthesis
Spondylolisthesis is a rare disorder that is most often associated with a pathologic luxation of the vertebra because of erosions of the pedicles or pars from foraminal neurofibroma or dural ectasia.
Tibial Dysplasia
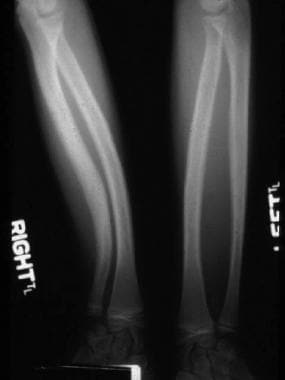
Tibial bowing occurs in 1 per 140,000 live births. The bowing associated with NF1 is always anterolateral. The deformity may appear before other protean manifestations, such as café-au-lait spots. It is usually evident within the first year of life, with a fracture not uncommonly occurring by the time the child is aged 2-2.5 years. Conversely, posteromedial congenital bowing, or kyphoscoliosis tibia, is a benign condition.
Tibial bowing associated with skin dimpling, ring constrictions, and foot deformities is rarely associated with NF1. Management of this anterolateral bowing deformity is most frustrating. Unlike scoliosis, treatment of congenital pseudarthrosis of the tibia does not appear to be more successful when it is initiated early.
There are two basic types of bowing: nondysplastic and dysplastic. Nondysplastic (type I) bowing is defined as follows:
-
Anterolateral bowing with increased bony density
-
Sclerosis of the medullary canal
-
Possibility of this type converting to dysplastic after fracture or osteotomy to correct the angulation
Dysplastic (type II) bowing is defined as follows:
-
Anterolateral bowing with a failure of tubulation
-
Anterolateral bowing with cystic prefracture or canal enlargement from previous fracture
-
Frank pseudarthrosis and atrophy with "sucked candy" narrowing of the ends of the two fragments
Spinal Treatment
Scoliosis
The treatment of nondystrophic curvatures is very similar to that of idiopathic scoliosis. Curves of less than 25° should be observed. Curves between 25° and 40° can occasionally be treated successfully with bracing. Once beyond 40°, surgery by posterior spinal fusion is usually indicated.
Dystrophic curvatures of less than 20° should be observed for progression at 6-month intervals. For persons with curvature greater than 20-40° of angulation, a posterior spinal fusion with some form of segmental spinal instrumentation is recommended. [16] Curves greater than 50° should be treated with anterior and posterior fusion.
Oblique radiographs are obtained every 6 months to exclude pseudarthrosis. Brace treatment has not been consistently effective.
For the very young child, early fusion causes minimal stunting of growth. Furthermore, the dystrophic segments have very limited growth potential to begin with. In these young children, nondystrophic curvatures of less than 20° should be observed, those of 20-35° should be braced, and those of 35° or greater should be stabilized by means of anterior and posterior fusion with segmental spinal instrumentation. [17]
Growing rods have been used in very young children with NF1 to obtain correction without definitive fusion and to lengthen or “grow the spine” every 6 months, but with varying success and a high rate of complications. [18] The MAGEC Spinal Bracing and Distraction System (NuVasive, Aliso Viejo, CA), which does not require operative intervention for lengthening, may be beneficial in managing the skeletally immature patient, but there is no current experience with this approach.
In a small study (N = 16) comparing posterior fusion (n = 8) with the growing-rod technique (n = 8) for early-onset NF1 dystrophic scoliosis, Cai et al reported no significant differences between the two groups with respect to the initial surgery correction rate or the rate of correction [19] ; however, the patients in the growth-rod group exhibited more T1-S1 growth during the follow-up overall and per year and had a significantly shorter operating time. The authors suggested that the growth-rod technique may be a more appropriate initial treatment but noted that the repeated procedures it requires may be a considerable disadvantage.
Kyphosis
Bracing is recommended for patients with kyphosis of less than 50°. In patients with curvatures greater than 50° that fail to correct on cross-table lateral x-rays taken hyperextended over a bolster, anterior surgery (intervertebral discectomy, rib strut grafting, and bone chip grafting) is required, followed by posterior segmental instrumentation. Once a curvature exceeds 70° and fails to correct as above, surgery is strongly indicated. Bracing is required after spinal surgery until there is evidence of bony fusion.
Because of the association of paraplegia with kyphosis, physicians have tended to perform laminectomy. Laminectomy alone for kyphotic cord compression is absolutely contraindicated. The offending neurofibromas are usually anterior, and decompression should be performed anteriorly. The removal of the posterior element predisposes the spine to instability by removing valuable bone stock required for fusion.
Spinal fusion should always be performed after laminectomy. A study by Helenius et al found that anteroposterior (AP) spinal fusion provided better correction of cervical kyphosis than posterior spinal fusion in children with NF1 and that fusion should include at least six levels to prevent junctional kyphosis. [20]
In a study comparing outcomes of anterior-only (AO), posterior-only (PO), and anteroposterior (AP) surgical approaches for treatment of dystrophic cervical kyphosis in patients with NF1, Lin et al found that the AP approach provided the best correction of dystrophic cervical kyphosis and sagittal balance and that patients treated in this manner were more satisfied with their outcomes. [21]
Lordoscoliosis
Anterior release and intervertebral fusion followed by posterior instrumented fusion is considered as the most reliable surgical option to achieve correction of dystrophic lordoscoliosis. Sublaminar wires, pedicle screws, Universal Clamps (Zimmer Spine, Bordeaux, France), or rod-multiple hook constructs can be used.
Spondylolisthesis
Anterior and posterior stabilization is recommended for progressive deformity. Progression of spondylolistheses from grade II to grade III or the presence of pathologically elongated pedicles with or without pain are indications for surgery. Fusion may be delayed because of the forward traction effect of the vertebral bodies and the slow healing and remodeling of bone in NF. Postoperative immobilization is indicated until the fusion is absolutely solid. Dural ectasia may completely erode the pedicles in this condition and continue to do so after a successful spinal fusion.
Considerations in Spinal Surgery
Subcutaneous plexiform neurofibromas are often overlying the incision area. When posterior spinal fusion surgery is performed, one should be aware of the very thin laminae, which are often eroded by dural ectasia surrounding the spinal cord in the thoracic region. The laminae, the pedicles, or both may be inadequate to accept hooks; pedicle screws, sublaminar wires, or Universal Clamps may be necessary. Considerable bleeding may occur with dissection around subcutaneous vascular tumors. The authors recommend using monopolar and bipolar electrocautery for subperiosteal exposure of the posterior elements.
Posterior subperiosteal dissection is performed by using Bovie electrocautery dissection rather than subperiosteal elevators because of the potential presence of laminar defects or the possibility of inadvertently plunging through the lamina and directly damaging the spinal cord.
Anterior spinal dissection may be complicated by venous lakes and engorgement of saccular, almost sinusoidal, vessels, which are difficult to control in and around the vertebral bodies. Extensive blood loss from the blood vessels in the cancellous bone of the vertebral bodies is distinctly possible. The authors recommend using a putty of Gelfoam (Pfizer, New York, NY), Avitene (Bard Davol, Warwick, RI), and thrombin. In addition, FloSeal (Baxter, Deerfield, IL) will assist in controlling bleeding, and Tisseel (Baxter) should be available for dural leaks.
Diskectomies should be performed with Bovie dissection and use of a rongeur through the anulus fibrosus instead of sharp dissections of the endplate apophysis. Sharp dissections may cause significant bleeding from the often-friable cancellous matrix of the vertebral bodies.
Rib head dislocation into the spinal canal should be ruled out by means of MRI before any attempt at surgical correction of the spinal deformity because correction may result in spinal cord impingement by the unrecognized dislocated rib head. Resection of the rib head is the most effective way of preventing the rib from being forced into the spinal cord during correction maneuvers.
Ileus is not infrequent when a staged anterior and posterior spine surgery is performed and while the patient is kept in craniofemoral traction. Therefore, preemptive nasojejunal tube placement and hyperalimentation are strongly recommended for all patients who undergo staged anterior and posterior surgery.
Progression of the deformity and erosion of bony elements may be noticed after solid spinal arthrodesis. Thus, close follow-up is needed even after successful spinal fusion.
Treatment of Tibial Dysplasia
Bracing can be both preventive and therapeutic. Children should be fitted with polypropylene knee-ankle-foot orthoses (KAFOs) as soon as they start to stand or walk around furniture. The KAFO is worn on a full-time basis, except during bathing.
Pulsating electromagnetic fields may also be used in the treatment of tibial dysplasia. The source of the fields may be external, such as the clamshell device over an ankle-foot orthosis (AFO), or an internal unit may be implanted in the soft tissue around the area of pseudarthrosis, usually in conjunction with an autogenous bone graft. The effectiveness of this form of treatment remains highly controversial.
Obvious documented fracture is the only current direct indication for surgery in congenital tibial dysplasia. In children older than 5 years, subsequent bracing above the knee and articulated at the knee and ankle has been tremendously successful in managing angular deformities and preventing fractures. The surgical treatments include direct bone grafting, vascularized autogenous grafting, compression and distraction osteogenesis, and amputation.
Subperiosteal dissection should be performed to expose the pseudoarthrosis site in congenital tibial dysplasia. Both ends are freshened, and a decision is made regarding whether an intramedullary rod or external fixation should be used. Use of an autologous iliac crest bone graft is highly recommended, regardless of the surgical procedure. A vascularized fibular bone graft is another option.
In following these patients, careful attention is paid to the ipsilateral distal fibular physis. If shortening occurs after a fibular fracture occurs or if the tibial operative procedure positions the distal fibular physis above the ankle joint, the foot and ankle will be in fixed valgus.
Surgical bone grafting
Autogenous bone is placed into the excised pseudarthrosis site. An intramedullary rod is placed from the proximal tibia across the pseudarthrosis site, incorporating the graft and extending down through the ankle, across the talus, and into the calcaneus. Rod replacement is required as the patient grows, and continuous bracing is necessary. A telescoping rod may be used and affixed to the distal tibia.
Another variation is to retrograde a rush rod from the calcaneus across the ankle, incorporating the grafted segment and continuing into the proximal tibial metaphysis, as described by Peter Williams. Most centers allow off-label use of bone morphogenetic protein (BMP) to support osteosynthesis.
The McFarland procedure, [22] placing a fibular allograft strut in the concavity of the bowed tibia, has been reintroduced into the options of bone grafting. It is usually performed as prophylaxis, before the fracture occurs.
Vascularized autogenous grafting
The most popular vascularized graft is the contralateral fibula, which is followed by the iliac crest. The graft is removed extraperiosteally and placed into the pseudarthrosis site. The blood vessels are then anastomosed to those normally supplying the tibia. Stabilizing the grafted segment is necessary. The distal tibia and fibula of the normal leg must be fused to prevent proximal migration of the fibular and ankle valgus.
Problems associated with vascularized grafts include progressive angular deformity, failure to achieve complete length, failure to unite with consequent further pseudarthrosis, valgus ankle instability, and disability in the donor limb.
Compression and distraction histogenesis
Compression and distraction histogenesis of bone and soft tissue using the Ilizarov method offers many theoretical advantages in the treatment of this problem. Unfortunately, the early glowing reports of bony synostosis failed in patients in whom bracing was not continued. Efforts now are being directed toward distraction and/or compression histogenesis over an intramedullary rod. Early reports have been encouraging. The Taylor spatial frame has replaced the Ilizarov as the external fixator of choice in some centers, but the technique is essentially the same. The system is more expensive.
Amputation
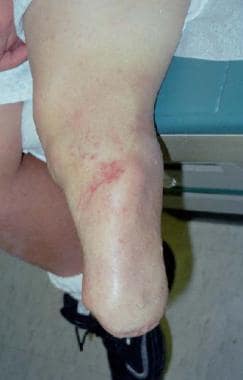
Amputation remains a viable alternative (see the image below). The weightbearing surface of the foot should be maintained á la Boyd-Syme amputation, as opposed to a transtibial amputation that predisposes the child to subsequent surgeries for bony stump overgrowth. The length from this procedure adds biomechanical stability to prosthetic wear. The new Seattle foot and/or runner's foot have made prosthetics more functional, and participation in team sports such as soccer is not prohibited after this procedure.
Surgical considerations
Regardless of the procedure used, some form of bracing is required for all of these patients until skeletal maturity is achieved. The potentially high incidence (25%) of subsequent central nervous system neoplasms should prompt consideration whether attempts at bone grafting should be continued after three or four procedures. By the time the child is aged 7 years, the benefit of further procedures, as opposed to amputation, should be scrutinized carefully.
Treatments for Disorders of Bone Growth
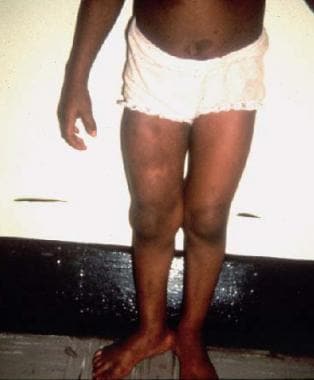
Two disorders of bone growth are segmental hypertrophy and subperiosteal proliferative bone growth. The diffuse hypertrophy of an extremity may be related to changes in the soft tissues (eg, hemangiomatosis, lymphangiomatosis, elephantiasis, and beaded plexiform neuromas), the combination of which is considered verrucous hyperplasia. (See the image below.)
The zones of overgrowth in bone and soft tissues are usually unilateral, involving the lower extremities or the head and neck. These osseous changes characteristically cause the bone to elongate with a wavy irregularity or thickening of the cortex. A higher incidence of neoplasia is associated with segmental hypertrophy than with other lesions.
Attempts to debulk the soft tissue and to resect the bone have not necessarily resulted in significant cosmetic improvement. Early epiphyseal arrest of the involved bone or leg lengthening of the normal, short side has shown improving results but is not perfect.
Treatment Contraindications
Spine
Bracing of progressive dystrophic curvatures is contraindicated simply because it has not been found to be effective. Some parents request bracing in spite of literature results questioning its efficacy. The authors' investigations have revealed a transition of idiopathic-type curves to dystrophic-type curves (modulation). Posterior spinal fusion alone is now believed to be contraindicated in young patients with progressive deformities. An anterior diskectomy and intervertebral fusion followed by posterior spinal fusion is the recommended procedure. The use of segmental fixation with instrumentation, as well as postoperative bracing, is highly recommended.
Limb-length inequality
Multiple attempts should be made to control growth in the hypertrophic limbs by means of epiphyseal arrest. A shortening osteotomy of the longer side is rarely successful and strongly contraindicated. The healing of the osteotomy may be delayed, and nonunion leading to pseudarthrosis may occur. Stapling of the longer limb has resulted in marginal successes, but a shortening osteotomy is strongly contraindicated. Lengthening the opposite side when the difference is less than 7 cm may be an alternative approach, but this is considered risky. [23]
Congenital tibial dysplasia
The elective correction of angular deformities for cosmesis is strongly contraindicated. The risk of pseudoarthrosis is too great. Consequent bracing and protection until skeletal maturity is highly recommended. [23] Skeletal maturity does not prevent fracturing, but in most instances, the patient becomes reasonable enough to refrain from taking the contraindicated risks.
-
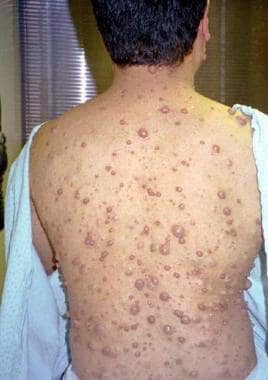
Multiple neurofibromas in 28-year-old man.
-
Plexiform neurofibroma of right thigh.
-
Radial and ulnar bowing and obliteration of intramedullary spaces.
-
Below-knee amputation for tibial pseudarthrosis.







