Practice Essentials
Mucopolysaccharidosis (MPS) involves defective activity of the lysosomal enzymes that degrade mucopolysaccharides (glycosaminoglycans [GAGs] attached to a link protein with a hyaluronic acid core) into smaller components. [1] The resulting incomplete degradation process leads to abnormal accumulation of heparan sulfate, dermatan sulfate, and keratan sulfate, and the abnormal accumulation of these compounds interferes with cell function.
Different forms of MPS were described separately throughout the 20th century. Their clinical presentations vary, depending on the type of enzyme defect and the glycoprotein accumulated. (See Lysosomal Storage Disease and Madelung Deformity.)
MPS can be subclassified as follows:
-
Hurler syndrome (MPS IH)
-
Hurler-Scheie syndrome (MPS I-H/S)
-
Scheie syndrome (MPS IS)
-
Hunter syndrome (MPS II)
-
Sanfilippo syndrome (MPS III)
-
Morquio syndrome (MPS IV)
-
Maroteaux-Lamy syndrome (MPS VI)
-
Sly syndrome (MPS VII)
In addition, MPS IX, an extremely rare form related to hyaluronidase deficiency, is recognized and is extremely rare. MPS X, caused by arylsulfatase K (ARSK) deficiency, has been proposed.
The image below depicts Morquio syndrome.
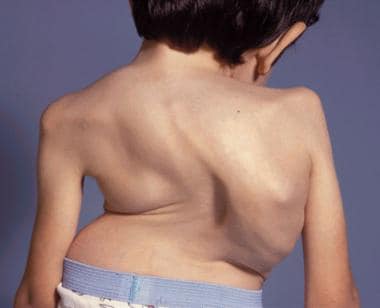 An 8-year-old boy with Morquio syndrome and severe kyphoscoliosis. Courtesy of Dennis P. Grogan, MD.
An 8-year-old boy with Morquio syndrome and severe kyphoscoliosis. Courtesy of Dennis P. Grogan, MD.
Signs and symptoms
Patients with MPS have normal development initially, with abnormalities appearing in infancy or later in childhood. Those with multiple organ system involvement may have the following presentations:
-
Central nervous system (CNS) disease – Hydrocephalus; cervical spine myelopathy
-
Cardiovascular disease – Angina; valvular dysfunction; hypertension; congestive heart failure
-
Pulmonary disease – Airway obstruction, potentially leading to sleep apnea, severe respiratory compromise, or cor pulmonale
-
Ophthalmologic disease – Corneal clouding; glaucoma; chronic papilledema; retinal degeneration
-
Hearing impairment – Deafness
-
Musculoskeletal disease – Short stature; joint stiffness; symptoms of peripheral nerve entrapment
Findings from examination may include the following:
-
MPS IH – Corneal clouding, hepatosplenomegaly, skeletal deformities (dysostosis multiplex), coarse facial features, large tongue, prominent forehead, joint stiffness, and short stature; upper-airway obstruction, recurrent ear infections, noisy breathing, and persistent nasal discharge; hirsutism, hearing loss, hydrocephalus, and intellectual disability
-
MPS I-H/S - Milder features; normal intelligence and micrognathia; corneal clouding, joint stiffness, and heart disease
-
MPS IS - Aortic valve disease, corneal clouding, and joint stiffness; normal intelligence and stature
-
MPS II (severe) – Pebbly ivory skin lesions on the back, arms, and thighs; coarse facial features, skeletal deformities, and joint stiffness; retinal degeneration with clear cornea and hydrocephalus, intellectual disability, and aggressive behavior
-
MPS II (mild form) – Similar features, but with much slower progression; normal intelligence and no hydrocephalus; hearing impairment and loss of hand function
-
MPS III – The most common MPS disorder; severe CNS involvement and only minimal somatic involvement; coarse hair, hirsutism, mild hepatosplenomegaly, and enlarged head; occasionally, mild dysostosis multiplex and joint stiffness; eventually, by age 8-10 years, profound intellectual disability with severely disturbed social behavior
-
MPS IV (severe) – Orthopedic involvement (eg, spondyloepiphyseal dysplasia) as the primary finding; preservation of intelligence; genu valgum, short stature, spinal curvature, odontoid hypoplasia, ligamentous laxity, and atlantoaxial instability
-
MPS IV (mild) – Much slower progression of skeletal dysplasia
-
MPS VI – Features very similar to MPS IH
-
MPS VII – Features similar to MPS IH
See Presentation for more detail.
Diagnosis
Antenatal screening studies that may be useful include the following:
-
Chorionic villus sampling
-
Amniocentesis
Postnatal diagnostic studies that may be helpful include the following:
-
Urinalysis, focusing on glycosaminoglycans (eg, dermatan sulfate, heparin sulfate, and keratin sulfate)
-
Serum assays for lysosomal enzymes (alpha-L-iduronidase, iduronate sulfatase, heparan N-sulfatase, N-acetylglucosaminidase, alpha-glucosamine- N-acetyltransferase, N-acetyl alpha-glucosamine-6-sulfatase, N-acetylgalactosamine-6-sulfatase, N-acetylgalactosamine-6-sulfatase, B-galactosidase, N-acetylgalactosamine-4-sulfatase, and B-glucuronidase)
Imaging studies that may be warranted are as follows:
-
Plain radiography (to detect dysostosis multiplex)
-
Computed tomography (CT) of the cranium (to help diagnose hydrocephalus)
-
Echocardiography (to monitor ventricular function and size in MPS patients with cardiovascular disease)
-
Magnetic resonance imaging (MRI; to evaluate spinal involvement)
Other tests to be considered are as follows:
-
Electroretinography
-
Audiologic assessment
See Workup for more detail.
Management
Specific treatment or cure is limited for MPS. Management has been limited to supportive care and experimental treatment modalities. Medical treatment modalities include the following:
-
Laronidase
-
Idursulfase
-
Elosulfase alfa
-
Vestronidase alfa
Surgical care for specific conditions may include the following:
-
Hydrocephalus – Ventriculoperitoneal shunting
-
Corneal clouding – Corneal transplantation
-
Cardiovascular disease – Valve replacement
-
Obstructive airway disease – Tracheostomy
-
Orthopedic conditions – Carpal tunnel release; soft-tissue procedures to release hip, knee, and ankle contractures; hip containment surgeries; corrective osteotomy for progressive valgus deformity at the knee; posterior spinal fusion
Multispecialty care is mandatory for these patients and should include a pediatrician (internist), a neurologist, a cardiologist, an ophthalmologist, an audiologist, an orthopedic surgeon, and a physical and occupational therapist.
See Treatment and Medication for more detail.
Pathophysiology
Defective activity of the lysosomal enzymes blocks the degradation process of mucopolysaccharides, leading to abnormal accumulation of heparan sulfate, dermatan sulfate, and keratan sulfate. These degradation by-products are then secreted and detected in the urine. MPS can be subclassified according to the type and amount of substance that accumulates, as follows [2, 3, 4] :
-
Hurler syndrome (MPS IH) (see the first three images below)
-
Hurler-Scheie syndrome (MPS I-H/S)
-
Scheie syndrome (MPS IS)
-
Hunter syndrome (MPS II)
-
Sanfilippo syndrome (MPS III)
-
Morquio syndrome (MP IV) (see the fourth through eighth images below)
-
Maroteaux-Lamy syndrome (MPS VI)
-
Sly syndrome (MPS VII)
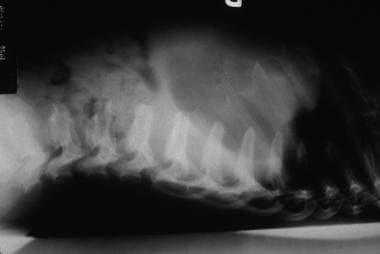 Hurler syndrome; lateral radiograph of thoracolumbar vertebrae illustrates vertebral plana. Courtesy of Bruce M. Rothschild, MD.
Hurler syndrome; lateral radiograph of thoracolumbar vertebrae illustrates vertebral plana. Courtesy of Bruce M. Rothschild, MD.
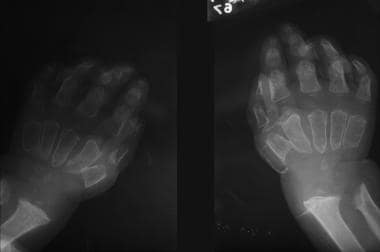 Hurler syndrome; widened metaphyses and diaphyses with truncated distal portions forming a peg characterize this radiograph. Courtesy of Bruce M. Rothschild, MD.
Hurler syndrome; widened metaphyses and diaphyses with truncated distal portions forming a peg characterize this radiograph. Courtesy of Bruce M. Rothschild, MD.
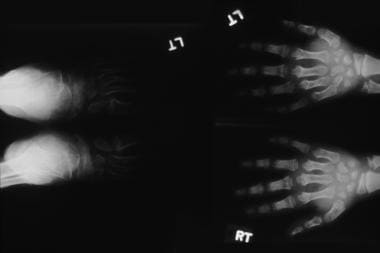 Hurler syndrome; widened metaphyses and diaphyses with truncated distal portions forming a peg characterize this radiograph. Courtesy of Bruce M. Rothschild, MD.
An 8-year-old boy with Morquio syndrome and severe kyphoscoliosis. Courtesy of Dennis P. Grogan, MD.
Hurler syndrome; widened metaphyses and diaphyses with truncated distal portions forming a peg characterize this radiograph. Courtesy of Bruce M. Rothschild, MD.
An 8-year-old boy with Morquio syndrome and severe kyphoscoliosis. Courtesy of Dennis P. Grogan, MD.
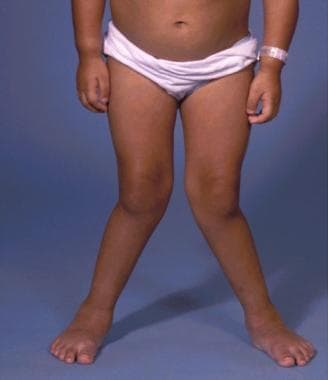 A 7-year-old girl with Morquio syndrome and typical severe genu valgum. Courtesy of Dennis P. Grogan, MD.
A 7-year-old girl with Morquio syndrome and typical severe genu valgum. Courtesy of Dennis P. Grogan, MD.
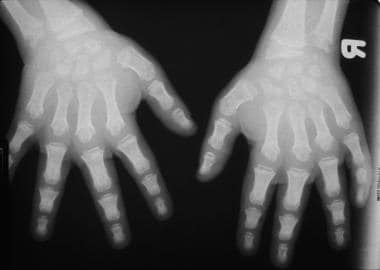 Morquio syndrome; widened bases of phalanges with osteopenia. Courtesy of Bruce M. Rothschild, MD.
Morquio syndrome; widened bases of phalanges with osteopenia. Courtesy of Bruce M. Rothschild, MD.
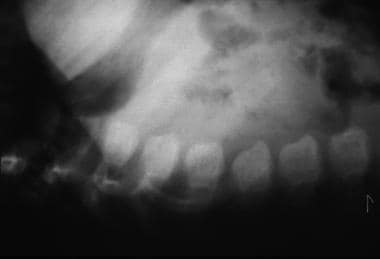 Morquio syndrome; lateral radiograph of thoracolumbar vertebrae illustrates vertebral body beaking. Courtesy of Bruce M. Rothschild, MD.
Morquio syndrome; lateral radiograph of thoracolumbar vertebrae illustrates vertebral body beaking. Courtesy of Bruce M. Rothschild, MD.
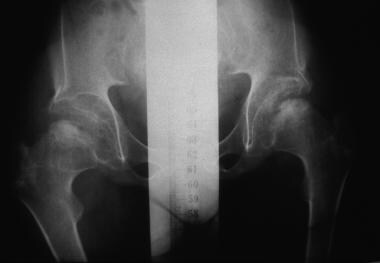 Morquio syndrome; anteroposterior radiograph of pelvis illustrates avascular necrosis of femoral head. Courtesy of Bruce M. Rothschild, MD.
Morquio syndrome; anteroposterior radiograph of pelvis illustrates avascular necrosis of femoral head. Courtesy of Bruce M. Rothschild, MD.
Another form of MPS, MPS IX, is related to hyaluronidase deficiency and is extremely rare. A European group proposed yet another form, MPS X, caused by arylsulfatase K (ARSK) deficiency. [5]
Etiology
All forms of MPS are inherited as autosomal recessive disorders, with the exception of Hunter syndrome (MPS II), which is inherited as a sex-linked recessive disorder.
Epidemiology
Worldwide, the prevalence of all types of MPS has been 1 case in 16,000-30,000 births. A study from Brazil estimated the birth prevalence of MPS to be 4.62 per 100,000 live births. [6] MPS III has accounted for 80% of cases.
The ages at which features of MPS present are somewhat variable. MPS features mostly present in the first few months of life. However, Morquio syndrome usually presents in children aged 2-4 years, and MPS IS and MPS VI can present late in childhood.
All MPSs are inherited as autosomal recessive disorders, with the exception of Hunter syndrome (MPS II); thus, all patients with Hunter syndrome are males.
These syndromes are found in all ethnic groups. The incidence of MPS II is higher in Israeli Jews, and the incidence of MPS IV is increased in French Canadians.
Prognosis
The prognosis varies, depending on the type of MPS. Most of these patients have shortened life spans, and some die in infancy. These disease processes have significant effects on the growth and development of the musculoskeletal system, including joint stiffness or hyperlaxity, deformities, and progressive loss of function. Multiple other organ systems are involved. The type and extent of organ system involvement are variable, depending on the subset of the disease.
Bone marrow transplantation has some positive effects systemically, such as reduction in hepatosplenomegaly, airway obstruction, and cardiopulmonary disease. These effects have resulted in improved life span, and many of these patients survive beyond the first decade of life.
-
An 8-year-old boy with Morquio syndrome and severe kyphoscoliosis. Courtesy of Dennis P. Grogan, MD.
-
A 7-year-old girl with Morquio syndrome and typical severe genu valgum. Courtesy of Dennis P. Grogan, MD.
-
Morquio syndrome; widened bases of phalanges with osteopenia. Courtesy of Bruce M. Rothschild, MD.
-
Morquio syndrome; lateral radiograph of thoracolumbar vertebrae illustrates vertebral body beaking. Courtesy of Bruce M. Rothschild, MD.
-
Hurler syndrome; lateral radiograph of thoracolumbar vertebrae illustrates vertebral plana. Courtesy of Bruce M. Rothschild, MD.
-
Morquio syndrome; anteroposterior radiograph of pelvis illustrates avascular necrosis of femoral head. Courtesy of Bruce M. Rothschild, MD.
-
Hurler syndrome; widened metaphyses and diaphyses with truncated distal portions forming a peg characterize this radiograph. Courtesy of Bruce M. Rothschild, MD.
-
Hurler syndrome; widened metaphyses and diaphyses with truncated distal portions forming a peg characterize this radiograph. Courtesy of Bruce M. Rothschild, MD.






