Background
First described by Koeppe, posterior polymorphous corneal dystrophy (PPMD) is a dominantly inherited condition characterized by particular alterations of the Descemet membrane and the corneal endothelium. [1] Typically, the corneal changes are either slowly progressive or nonprogressive. In severe cases, corneal decompensation and edema can occur. Although PPMD is most often a bilateral condition, marked asymmetry in the degree of involvement may be seen. Most persons with PPMD are asymptomatic.
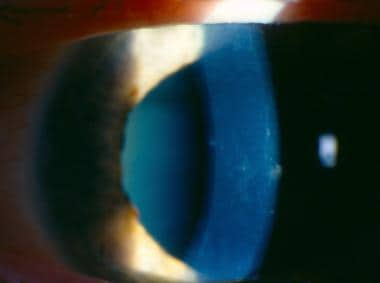 Slit lamp image demonstrates posterior corneal vesicles and opacities in linear bands and other polymorphous configurations typical of posterior polymorphous corneal dystrophy.
Slit lamp image demonstrates posterior corneal vesicles and opacities in linear bands and other polymorphous configurations typical of posterior polymorphous corneal dystrophy.
Pathophysiology
Three main abnormalities are described: vesicular changes, endothelial band lesions, and irregular diffuse opacities of the posterior corneal surface involving both the Descemet membrane and the endothelium. The corneal endothelium undergoes a transformation and demonstrates many epithelial characteristics on examination with electron microscopy and immunohistochemical analysis. Often, the endothelium is found to be multilayered. Most patients exhibit vesicular lesions, which are the hallmark of PPMD.
Epidemiology
Frequency
United States
The frequency of PPMD is not well documented. Although it is considered to be uncommon, PPMD may be recognized and diagnosed more often in recent years than in the past.
International
Liskova et al found that the prevalence of PPCD in the Czech Republic appears to be the highest worldwide. [2]
Mortality/Morbidity
The effect of PPMD on patients is highly variable, with a broad clinical spectrum of findings, ranging from nonprogressive asymptomatic disease to progressive or advanced debilitating corneal disease with corneal decompensation and glaucoma.
Race
No racial predilection exists.
Sex
No sexual predilection exists.
Age
Although PPMD is an autosomal dominantly inherited corneal dystrophy, the age at diagnosis is highly variable because of the broad spectrum of disease severity. Findings may be present at birth. Most patients are first identified at age 30-50 years; however, this is likely only indicative of a more common age for ocular examinations.
Some patients may present at birth with congenital disease, exhibiting advanced disease with corneal edema.
Presentation in adulthood is indicative of a more stable disease state with a decreased probability of progression to corneal decompensation.
Most patients with significant symptoms present at age 25-50 years.
-
Slit lamp image demonstrates posterior corneal vesicles and opacities in linear bands and other polymorphous configurations typical of posterior polymorphous corneal dystrophy.

