Overview
Multifocal choroidopathy syndromes are rare disorders involving a primary pathologic process occurring at or near the level of the retinal pigment epithelium (RPE) with or without photoreceptor outer segment and choriocapillaris involvement. Ultimately, the etiology is presumed to be either a vasculitic obstruction of the choriocapillaris with secondary infarction of the overlying RPE or an immunologic response directed at the RPE itself. Many clinical features of the individual syndromes overlap, causing much confusion. Whether these conditions represent distinct entities or parts of a spectrum of the same basic disease is still unknown.
Other conditions of choroiditis with overlying RPE involvement are not described in this article; yet, they are important to consider in the differential diagnosis of the more atypical choroiditides, such as tuberculosis, histoplasmosis, syphilis, posterior scleritis, reticulum cell sarcoma, lymphoid neoplasia, diffuse unilateral subacute neuroretinitis, and Lyme disease, or the more common idiopathic syndromes, such as the pachychoroidal disease spectrum, sarcoidosis, Vogt-Koyanagi-Harada syndrome, and sympathetic ophthalmia.
Classification
The earliest classification of multifocal choroidopathy syndromes, established by Gass, [1] describes the acute zonal occult outer retinopathy (AZOOR complex) spectrum of diseases. This encompasses multiple evanescent white dot syndrome (MEWDS), acute idiopathic blind spot enlargement syndrome (AIBSES), acute macular neuroretinopathy, presumed ocular histoplasmosis, punctate inner choroidopathy, and multifocal choroiditis. [1] The basis for this classification was that these disorders occurred primarily in young adult women, affected the outer retina and choroid, and were associated with inflammation, visual field loss, and in some instances, electroretinogram abnormalities. [2] Subsequently, Cimino and colleagues [3] suggested an ischemic choroidopathy, which resulted from primary inflammation of the choriocapillaris, as the unifying pathogenesis of these diseases.
The AZOOR complex should be differentiated from well-defined AZOOR. Although this complex of disorders could manifest as or precipitate AZOOR, in most patients AZOOR does not develop. Although the presentation of AZOOR can vary, there are some common clinical features.
On funduscopic examination, minimal initial ophthalmoscopic changes are seen. One common finding is a grayish-white line that demarcates the normal and involved retina. Within weeks, this line is replaced by an orange zone, which eventually becomes a larger sector of retinal pigment epithelium (RPE) depigmentation and retinal vasculature attenuation. An afferent pupillary defect is present in 75% of patients, along with enlarging visual field scotomas. [4, 5] Mrejen and colleagues [6] have described clinical fundus and multimodal imaging findings which are unique to AZOOR.
Since then, AIBSES has been characterized to have sufficiently unique clinical features that warrants its classification as a separate entity. [7, 8] Acute macular neuroretinopathy, a bilateral condition affecting otherwise healthy young adults, appears to involve a pathologic process occurring more in the middle and outer retinal layers rather than in the RPE and choriocapillaris. [9] Therefore, AIBSES and AMN are not discussed in this article.
Outside of similar presenting symptoms and some overlapping clinical features, multifocal choroidopathy syndromes lack any significant histopathologic feature, serologic finding, or common attributable etiology. [10] Accordingly, discussion of the various multifocal choroidopathy syndromes in this article addresses each disease as a separate entity.
Multifocal Choroiditis
Multifocal Choroiditis and Panuveitis
First described by Nozik and Dorsch in 1973, [11] and later, by Dreyer and Gass [12] and Deutsch and Tessler, [13] multifocal choroiditis and panuveitis (MCP) has features that superficially resemble primary ocular histoplasmosis (POHS), with the additional findings of aqueous, vitreous and chorioretinal inflammation.
Historically, punctate inner choroidopathy (PIC) has been described as chorioretinal lesions of the posterior pole in young myopic women without ocular inflammation; however, in many peer-reviewed studies, this disorder is considered to be a variant of MCP. Spaide and colleagues [14] demonstrated no differences in multimodal imaging between MCP and PIC. Accordingly, this article addresses the two as a single disease entity. [14]
Multifocal choroiditis and panuveitis affects otherwise healthy young patients in the third to fourth decade, with an incidence of bilateral involvement of 80%. [15] A moderate racial and sex predisposition exists, with approximately 66% of patients being White and 80% of patients being female. [15] Myopia is also present in more than 85% of these individuals.
Signs and symptoms
The principle presenting symptoms of MCP are usually blurry vision, with or without photophobia, and scotomas.
Presenting visual acuity may vary from 20/20 to light perception, with an average of 20/100. Decreased visual acuity is present in about 70% of patients. Clinical examination reveals mild-to-moderate aqueous inflammation in approximately 50% of patients and vitreous inflammation in 90% of patients. In patients with minimal anterior chamber/vitreous inflammation, the mean presenting visual acuity is 20/46. [16] Of note, the anterior chamber and vitreous inflammation is paroxysmal, and may not be observed during a particular clinic visit. [17]
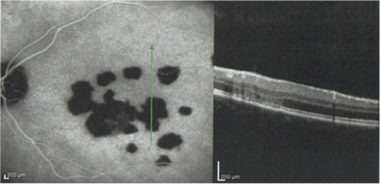
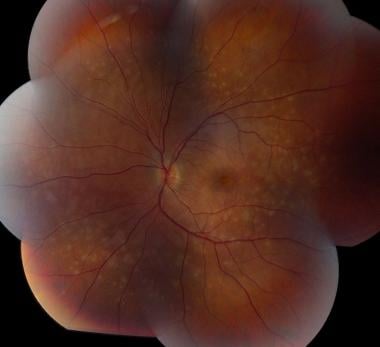
On funduscopic examination, the acute phase is characterized by multiple yellowish, round/oval lesions clustered at the macula and optic nerve. They often extend to the mid-periphery or beyond and assume a a linear configuration, called Schlaegel lines. With time, these lesions may lead to chorioretinal scarring, or less commonly, subretinal fibrosis. [18] Other vision-affecting associated signs are cystoid macular edema, optic disc edema, and choroidal neovascular membrane. [19]
These lesions are approximately 200 µm in diameter, but they can vary in size (range, 50-1000 µm). The lesions tend to scatter predominantly in the periphery rather than in the macula and are located at the level of the retinal pigment epithelium (RPE) and choriocapillaris. Multiple active lesions may occur at any one time.
During the late stage of the disease, the spots may become atrophic, with a rim of hyperpigmentation. These can assume the classic punched-out appearance with bands of subretinal fibrosis at their margins.
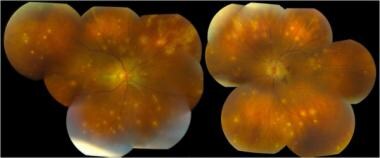 Multifocal choroiditis and panuveitis (early stage): Multiple, white, discrete lesions at the level of the choroid. Courtesy of the Journal of Ophthalmic Inflammation and Infection, BioMed Central Ltd, Springer Nature.
Multifocal choroiditis and panuveitis (early stage): Multiple, white, discrete lesions at the level of the choroid. Courtesy of the Journal of Ophthalmic Inflammation and Infection, BioMed Central Ltd, Springer Nature.
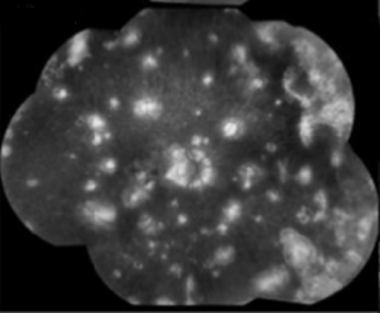
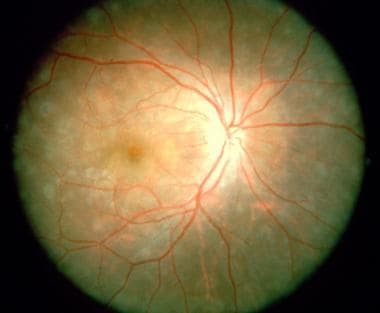
 Late stages of multifocal choroiditis: Note the multiple atrophic lesions. Courtesy of the Case Reports in Ophthalmological Medicine journal, Hindawi.
Late stages of multifocal choroiditis: Note the multiple atrophic lesions. Courtesy of the Case Reports in Ophthalmological Medicine journal, Hindawi.
Diagnostic imaging
Optical coherence tomography (OCT) reveals diffuse ellipsoid zone disruption beyond the chorioretinal lesions. Autofluorescence imaging studies may show hyperfluorescent and hypofluorescent patches that correspond to visual field defects. [20]
During the early stage of MCP, the active lesions exhibit blockage of the early fluorescence followed by late hyperfluorescence. The later (inactive) stage of the disease exhibits RPE window defects corresponding to the areas of the atrophic lesions. [21]
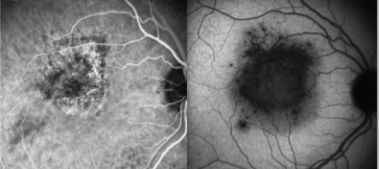
 Early disease stage (top): Fluorescein angiography reveals discrete, roundish hypofluorescent lesions, which are clinically unapparent, but correspond to future lesions. Late disease stage (bottom): Hypofluorescent lesions have considerably increased in number and are hyperfluorescent in late phase of the fluorescein angiography. A discrete macular late leakage is found in the right eye consistent with choroidal neovascularization. Courtesy of the Journal of Medical Case Reports, BioMed Central Ltd, Springer Nature.
Early disease stage (top): Fluorescein angiography reveals discrete, roundish hypofluorescent lesions, which are clinically unapparent, but correspond to future lesions. Late disease stage (bottom): Hypofluorescent lesions have considerably increased in number and are hyperfluorescent in late phase of the fluorescein angiography. A discrete macular late leakage is found in the right eye consistent with choroidal neovascularization. Courtesy of the Journal of Medical Case Reports, BioMed Central Ltd, Springer Nature.
 Multifocal choroiditis: Late phase of fluorescein angiogram showing staining of inactive lesions. Courtesy of the Journal of Clinical and Experimental Ophthalmology, Longdom Publishing.
Multifocal choroiditis: Late phase of fluorescein angiogram showing staining of inactive lesions. Courtesy of the Journal of Clinical and Experimental Ophthalmology, Longdom Publishing.
The early lesions of MCP are patchy and hypocyanescent on indocyanine green (ICG) angiography, with persistence and expansion of hypocyanescence in the late phase.
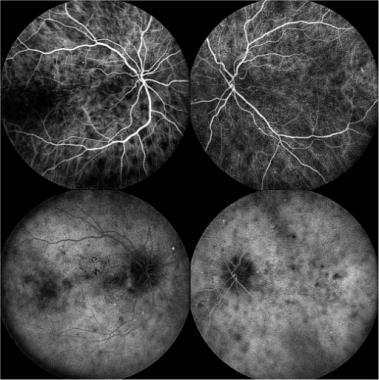 Early phase ICG (top) and Late phase ICG (bottom): The lesions remained hypocyanescent throughout all phases of the angiogram
Early phase ICG (top) and Late phase ICG (bottom): The lesions remained hypocyanescent throughout all phases of the angiogram
Electrophysiologic and psychophysical studies
Findings on the electroretinogram (ERG) are normal in approximately 50% of patients with MCP. It has been reported that the acute zonal occult outer retinopathy (AZOOR) complex diseases share a pattern of visual dysfunction that originates in the photoreceptors, has a patchy distribution across the retina, and is asymmetric in both eyes. [22]
The electrooculographic (EOG) findings are abnormal in approximately 60% of patients studied. Visual field testing sometimes demonstrates increased blind spot size. [23] Visual field defects correspond to patches of active retinal disease seen on autofluorescence. Visual field pattern abnormalities correlate well with ERG a-wave amplitude. [2]
Etiology
The etiology of MCP remains unknown. Extensive systemic medical evaluations have not identified a single common cause for MCP. In contrast to 90% of patients with POHS who exhibit positive reactions to the histoplasmin skin test, only 25% of patients with MCP exhibit positive reactions to this skin test. [12] Positive treponemal serology is evident in up to 27% of patients with MCP, yet no modification of the disease with penicillin treatments has been achieved to date.
The presumed causes of multifocal choroidopathy (MFC) lesions can be classified as infectious (POHS, tuberculosis, brucellosis, coccidiomycosis, candidiasis and other fungal septicemias, syphilis, West Nile virus, Zika etc), [24, 25, 26, 27, 28, 29] noninfectious (sarcoidosis and other granulomatous diseases [eg, Blau syndrome]), and idiopathic. [30]
According to Spaide and colleagues, [14] the principle sites involved appear to be the sub-RPE and outer retinal spaces. Acute lesions are often associated with more widespread involvement of the ellipsoid zone (EZ) extending beyond the zones of RPE involvement.
Disease course and prognosis
Idiopathic MCP is a chronic progressive bilateral disease. One of the most common causes of visual loss in patients with MCP is related to development of choroidal neovascular membranes. [31] The visual prognosis is variable, more favorable in patients in whom the inflammation is confined to the retina (ie, multifocal choroiditis without panuveitis) or in those where the active retinal lesions do not threaten the fovea. [16]
Unfortunately, in patients with more extensive or diffuse chorioretinal atrophy, the visual prognosis remains poor. [32, 33, 34] Peripapillary atrophy with progressive subretinal fibrosis results in enlargement of a blind spot in approximately 43% of patients. [23, 35] Approximately one-third of patients with MCP maintain their initial visual acuity at onset, whereas another one-third lose, on average, at least 2 or more Snellen lines. [32]
Treatment
In cases of vision-threatening choroidal neovascularization, anti-vascular endothelial growth factor (anti-VEGF) therapy predominates; however, in the absence of CNV, there is no universally accepted treatment. Most patients can be monitored without treatment, unless active lesions are threatening the fovea. In patients with active lesions, first-line therapy consisting of systemic corticosteroids has been shown to improve visual outcomes. [20]
In recurrent cases, immunomodulatory therapy (eg, azathioprine, cyclosporine, or methotrexate) has showed promising results in controlling inflammation and preserving of vision. [36]
Diffuse Subretinal Fibrosis Syndrome
Diffuse subretinal fibrosis uveitis (DSFS) syndrome is a disease on 1 end of the multifocal choroiditis (MFC) spectrum. Specifically, enlarging and coalescing areas of subretinal fibrosis eventually involve the macular involvement and lead to visual loss. DSFS carries a poor visual prognosis despite use of high-dose corticosteroids and systemic immunosuppression. The affected demographic is healthy individuals in the second decade of life (average age, approximately 20 years; range, 14-34 years). [37]
Signs and symptoms
Patients typically present with central visual loss and reports of metamorphopsia. Clinical examination during the acute phase reveals multiple, small, yellow stellate lesions scattered throughout the posterior pole. Mild vitritis may be present with isolated pockets of subretinal fluid. In the chronic phase, the lesions become discrete with sharply angulated subretinal scar formation. In the macula, the lesions tend to coalesce, forming broad zones of subretinal fibrosis. Later complications can include serous and hemorrhagic macular detachment.
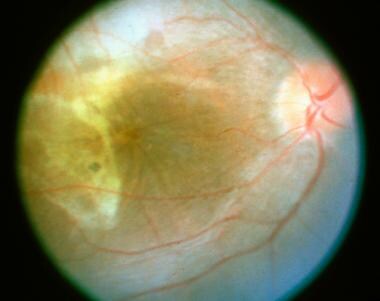 Diffuse subretinal fibrosis syndrome. This patient's right eye shows typical subretinal angulated scar formation.
Diffuse subretinal fibrosis syndrome. This patient's right eye shows typical subretinal angulated scar formation.
Diagnostic testing
As mentioned earlier, this condition shares many clinical features with classic MCP. Although the findings on angiograms are similar, the distinct features of DSFS are an early-phase hypofluorescence in the areas of subretinal fluid loculation. During the late phase of the angiogram, extensive leakage is present at these sites, which in some cases, may suggest subretinal neovascularization. [38]
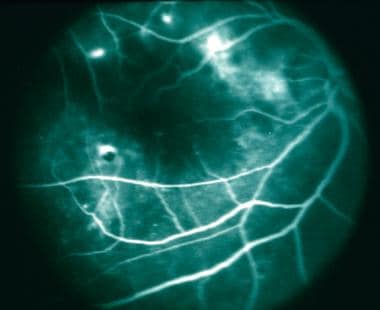 Diffuse subretinal fibrosis syndrome. Midphase of fluorescein angiogram demonstrating progressive leakage hyperfluorescence.
Diffuse subretinal fibrosis syndrome. Midphase of fluorescein angiogram demonstrating progressive leakage hyperfluorescence.
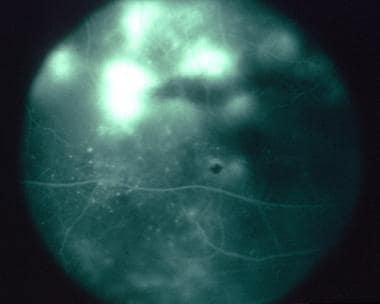 Diffuse subretinal fibrosis syndrome. Late phase of the fluorescein angiogram demonstrating progressive leakage as hyperfluorescence.
Diffuse subretinal fibrosis syndrome. Late phase of the fluorescein angiogram demonstrating progressive leakage as hyperfluorescence.
Electrophysiologic and psychophysical studies
The findings on the electroretinography and the electrooculography are variable in this condition, ranging from intact to severely depressed function. Visual field testing reveals constriction peripherally with central scotomas that often correlate with the clinical lesions.
Etiology
Like MFC, DSFS has no associations with systemic disease. Some case reports have linked DSFS to hormonal irregularities in young women and even energy drink consumption. [39, 40] In view of the clinical and angiographic findings, this condition's clinical appearance is likely secondary to a primary RPE hyperplastic process. [41] Histologic evidence of immunoglobulin and complement deposition on the internal side of the Bruch membrane also suggests a focal inflammation of the RPE. [42]
Disease course and prognosis
This condition is characterized by recurrent episodes with the fellow eye becoming involved within a 6-month period. Visual prognosis is generally poor.
Management of DSFS involves the use of corticosteroids early in the course of the disease. [43] This treatment is useful in preventing significant loss of vision in the contralateral eye. Immunomodulators may be useful when corticosteroids have failed to control the disease.
Acute Posterior Multifocal Placoid Pigment Epitheliopathy
In 1968, Gass [44] first described acute posterior multifocal placoid pigment epitheliopathy (APMPPE). This condition typically affects young or middle-aged, otherwise healthy, adults (average age, 25 years; range, 7-57 years) who experience transient acute central or paracentral vision loss. [45, 46] There is no racial nor sex predilection; however, a recent 20-year study of the incidence of white dot syndromes in Olmstead County, Minnesota found that multiple evanescent white dot syndrome (MEWDS) occurred more commonly in females, and APMPPE occurred more often in males. [47] Bilateral involvement is generally the rule, although involvement of the second eye may be delayed by several weeks.
The condition tends to resolve spontaneously in approximately 7 to 11 weeks with return of near-normal visual acuity and visual function in most patients. Although some retinal pigment epithelium (RPE) changes may remain visible and small paracentral scotomas may persist, no other ocular disturbances are usually evident. The typical course of the disease suggests an underlying viral inflammatory disorder [1, 48] or a delayed type IV hypersensitivity reaction to multiple stimuli. [49]
Of note, it has been discovered that APMPPE may represent a manifestation of primary central nervous system (CNS) vasculitis. Given the high mortality rate associated with CNS vasculitis, neuroimaging and consultation with a neurologist are warranted for these patients. [50]
Signs and symptoms
A rapid reduction in visual acuity typically occurs with or without a history of a preceding flulike illness or upper respiratory tract infection (present in 33%). [51] Acute posterior multifocal placoid pigment epitheliopathy presents clinically as multiple whitish-yellow inflammatory lesions at the outer retina, retinal pigment epithelium, and choroid. [15]
Lesions in APMMPE lesions have a size of 125 μm, assume a placoid conformation, and are associated with minimal vitreous inflammation. The retina overlying the lesions appears normal. Lesions are typically scattered throughout the posterior pole, including the macula, and in the postequatorial midperiphery. Initially, they appear cream-colored or grayish-white in appearance.
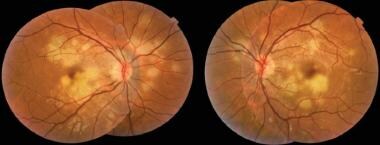 Color fundus photography images show multiple bilateral yellow-white placoid lesions, at the level of external retina, posterior to the equator, and a slight fade of the edges of the optic nerve. Courtesy of the Case Reports in Ophthalmological Medicine journal, Hindawi.
Color fundus photography images show multiple bilateral yellow-white placoid lesions, at the level of external retina, posterior to the equator, and a slight fade of the edges of the optic nerve. Courtesy of the Case Reports in Ophthalmological Medicine journal, Hindawi.
Anterior segment inflammation is usually not observed, although some minimal vitreous inflammation may be present. Signs of vascular inflammation (periphlebitis) and mild optic disc edema may also be noted. Very rarely, the condition can result in retinal edema, retinal hemorrhage, and choroidal neovascular membrane formation. [52]
Diagnostic imaging: fundus autofluorescence
During the active phase, the lesions demonstrate hypoautofluorescence with hyperautofluorescent margins. [53, 54, 55, 56] Hypoautofluorescence of the fovea on fundus autofluorescence may correlate with visual acuity impairment in the white dot syndromes, including APMPPE. [57]
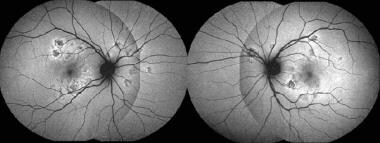 Fundus autofluorescence: Hypoautoflourescence corresponding to the placoid lesions, with hyperautofluorescent edges. Courtesy of the Case Reports in Ophthalmological Medicine journal, Hindawi.
Fundus autofluorescence: Hypoautoflourescence corresponding to the placoid lesions, with hyperautofluorescent edges. Courtesy of the Case Reports in Ophthalmological Medicine journal, Hindawi.
Diagnostic imaging: fluorescein angiography
The initial phase of the angiogram sometimes demonstrates prolonged filling of the choroidal vasculature. The acute-phase lesions are hypofluorescent. This early hypofluorescence is often attributed to a combination of incomplete and irregular filling of the choriocapillaris in conjunction with a masking effect produced by the edematous, swollen RPE cells. This is followed in the late phase with evidence of leakage and staining at the lesion sites.
The healed lesions of APMPPE demonstrate mottled hyperfluorescence and window-type defects caused by alterations in the RPE in areas corresponding to the acute lesions.
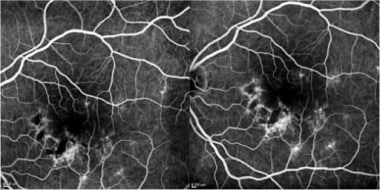 Fluorescein angiogram in a patient with APMPPE: Early (left) and intermediate (right) phases show hyperfluorescence with moderate staining of lesions.
Fluorescein angiogram in a patient with APMPPE: Early (left) and intermediate (right) phases show hyperfluorescence with moderate staining of lesions.
Diagnostic imaging: indocyanine green angiography
The typical ICG pattern for both active and inactive lesions consists of hypofluorescent areas up to the late phase, suggesting choriocapillaris nonperfusion. [58, 59, 60, 61]
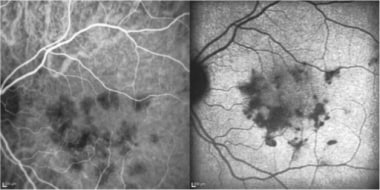 Early phase (left) and late phase (right) frames reveal more extensive areas of involvement (hypofluorescence) than seen on clinical examination.
Early phase (left) and late phase (right) frames reveal more extensive areas of involvement (hypofluorescence) than seen on clinical examination.
Diagnostic imaging: optical coherence tomography scanning
Initially, the active phase of the disease demonstrates an anterior displacement and hyperreflectivity of neuroretina and outer reflective bands in areas overlying affected RPE. [62, 54, 56, 63, 64, 65] A spectral domain optical coherence tomography (SD-OCT) study of a patient with acute APMPPE lesions, however, revealed a hyperreflective area located between the outer nuclear layer and the inner and outer photoreceptor segments (inner segment-outer segment [IS/OS] junction) corresponding to the placoid lesions seen clinically and angiographically. [66] This finding suggests that the whitish placoid lesions seen clinically in patients with APMPPE may represent edema between the outer nuclear layer and the IS/OS junction rather than swelling or opacification of the RPE cells.
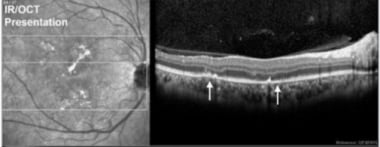 Note the disruption of RPE/outer retina that corresponds to placoid lesions (white arrows). Courtesy of the Journal of Clinical and Experimental Ophthalmology, Longdom Publishing.
Note the disruption of RPE/outer retina that corresponds to placoid lesions (white arrows). Courtesy of the Journal of Clinical and Experimental Ophthalmology, Longdom Publishing.
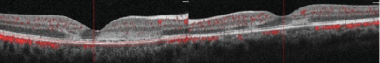 SD-OCT: Note the hyperreflectivity from the outer plexiform layer to the RPE. The retina is of normal thickness. Courtesy of the Case Reports in Ophthalmological Medicine journal, Hindawi.
SD-OCT: Note the hyperreflectivity from the outer plexiform layer to the RPE. The retina is of normal thickness. Courtesy of the Case Reports in Ophthalmological Medicine journal, Hindawi.
Goldenberg and colleagues [67] have proposed a 4-stage classification system for APMPPE based on SD-OCT findings in a study of 12 eyes of 6 patients.
Stage 1a, the hyperacute phase of the placoid lesions, is characterized by a well-demarcated dome-shaped elevation of the IS/OS junction with hyperreflective intertwined material and variable amounts of subretinal fluid present beneath the IS/OS junction and the RPE. Stage 1b, the acute phase lesion, is characterized by a fairly rapid flattening of the well-demarcated dome-shaped elevation accompanied by thickening of the IS/OS junction and hyperreflectivity of the outer nuclear layer.
Within about 2 weeks, the lesions evolve into stage 2, a subacute phase, and a distinct separation of the IS/OS junction and RPE layer is visible with mild subretinal fluid accumulation present. The hyperreflectivity of the outer nuclear layer begins to fade, and the layer thins at this stage.
In stage 3, the late phase, which occurs approximately 6 weeks after disease onset, there is partial disappearance of the IS/OS junction, and accentuated hyperreflectivity of the RPE is seen.
At 3 months after onset, stage 4, or the resolution phase, is visible. In the resolution phase, 2 hyperreflective bands, indicative of the IS/OS junction and the RPE, reappear as separate and distinct bands.
In some instances, on OCT, APMPPE may have morphologic OCT features similar to those of Vogt-Koyanagi-Harada disease, notably, subretinal fluid with multilobular spaces and marked choroidal thickening. [68]
Electrophysiologic and psychophysical studies
Most patients with APMPPE have normal EOG findings; however, some patients may have diminished, subnormal EOG findings during the acute phase of the disease. Some may exhibit subnormal cone and rod ERG findings. Scotomas on visual-field testing are often present during the acute phase of the disease, which may or may not be coincident with the observable clinical lesions. These scotomas can persist and be permanent.
Etiology
The pathogenesis of APMPPE is not entirely understood; however, it seems that the primary insult is at the level of inner choroid with retinal changes occurring secondarily. [69, 70] It has been postulated that the cause is an obstructive vasculitis, which results in ischemia of terminal choroidal lobules in the posterior pole. The overlying RPE changes are secondary to this ischemic injury. [68] Accordingly, some authors have suggested that this entity should be renamed to reflect its true nature as a choroidopathy. [71]
Of all the conditions described in this article, APMPPE has the greatest variety of associations with other systemic conditions and complications. The systemic conditions are as follows [55, 72, 73, 74, 75, 76, 77, 78, 79, 80] :
-
Platelet aggregation abnormalities
-
Lymphadenopathy
-
Renal cell carcinoma [74]
-
Systemic mycobacterial infections [82]
-
Hepatomegaly
-
Regional enteritis
-
Recurrent stroke [87]
-
Recurring meningoencephalitis [90]
-
Cranial nerve VI paralysis [91]
-
Vitamin B-12 deficiency [77]
-
Homocysteinemia [77]
-
Microvascular nephropathy [94]
-
Drug Reaction with Eosinophilia and Systemic Symptoms syndrome [95]
-
Hemophagocytic syndrome [80]
-
Dysacusis [96]
-
Adenovirus type V [76]
-
Reaction to swine influenza Vaccine [72]
-
Reaction to influenza vaccination [97]
-
Spinal fluid pleocytosis and elevated cerebral spinal fluid (CSF) protein
-
Adverse effect of alemtuzumab treatment [98]
The association between APMPPE and cerebral vasculitis is well known. Given the morbidity and mortality rates associated with CNS vasculitis, especially when it occurs after a rapid taper of systemic steroids, neurologic evaluation is often warranted. [66, 99, 100, 101] Furthermore, any systemic symptom should prompt the clinician to evaluate for other CNS and systemic granulomatous diseases.
Disease course and prognosis
The placoid lesions in APMPPE spontaneously resolve in most cases, and visual acuity is usually recovered within a few months. [102] The acute lesions usually begin to fade after a month, leaving pigment mottling and atrophic, geographic-shaped lesions at the level of the RPE. Improvement in visual acuity often lags the resolution of the RPE lesions, which can take from weeks to months. Fortunately, the prognosis for visual recovery is very good, and many patients (75-80%) experience better than 20/40 vision. [103, 104] Some eyes remain symptomatic, however, and visual acuity remains reduced in up to 40% of patients if there is foveal involvement at presentation. [104] Recurrences, unlike other conditions covered in this section, are rare, and are usually experienced within the first 6 months. [105] If initial presentation is unilateral, the second eye is often affected a few days or weeks after the first eye.
Treatment
Visual outcome is normally good, with more than 80% of patients who have APMPPE without foveal involvement experiencing resolution of visual acuity of 20/25 or better, but some patients experience incomplete restoration of visual function. [51] Accordingly, therapy with corticosteroids may have a role when any of the following conditions are present: foveal involvement, advanced age, unilateral disease, recurrence, or risk for CNS vasculitis. [106] A report of APMPPE lesions treated with an intravitreal dexamethasone implant demonstrated restoration of the ellipsoid zone on OCT.
Other treatments, such as the tumor necrosis factor blocker infliximab have been reported to improve visual acuity and prevent recurrences during the follow-up period [107] ; however, treatment with infliximab did not prevent disease spread to the contralateral eye. Sub-Tenon injections and intravitreal triamcinolone have been used for associated complications, such as macular edema and choroidal neovascularization. [108, 109, 110]
Acute Retinal Pigment Epitheliitis (Krill Disease)
Acute retinal pigment epitheliitis (ARPE) is a very rare condition, first described by Krill and Deutman [111] in 1972. Acute retinal pigment epitheliitis is a self-limited disease of the retinal pigment epithelium (RPE) that typically occurs in young, otherwise healthy adults. The median age at onset is 45 years, with a range of 16-75 years. A preponderance of involvement is evident in males (approximately two-thirds). No racial predilection exists, and the condition is bilateral in approximately 40% of patients.
Signs and symptoms
Approximately half of patients have symptoms at the time of presentation and exhibit the characteristics of acute central scotoma and fine pigment stippling in the macula surrounded by hypopigmented halos. [112] The lesions appear dark gray to black, with surrounding yellow-white halos; they typically measure 50-100 µm. Often, 1 to 4 golden spots are arranged in discrete clusters around the macula. With resolution, the spots may either darken or lighten, but the halos remain. Extramacular lesions are rare, and, with time, a pattern of RPE migration may develop at the sites of the spots. Usually, there is no associated ocular inflammation. [113, 114]
Diagnostic imaging: fluorescein angiography
During the acute phase of the disease, the centers of the spots are hypofluorescent as a result of blockage. With disease progression and ensuing RPE changes, hyperfluorescence caused by a window defect becomes apparent. [115]
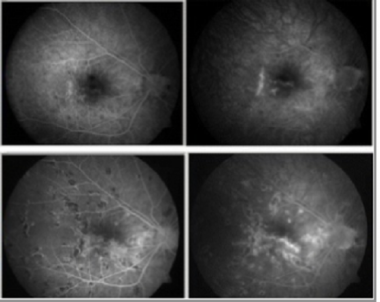
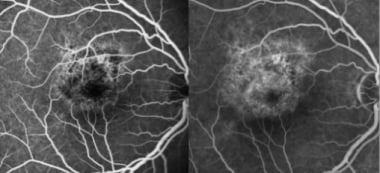 Acute retinal pigment epitheliitis: Early-phase fluorescein angiography (left) vs late-phase fluorescein angiography (right). Courtesy of the Clinical Ophthalmology journal, Dove Medical Press.
Acute retinal pigment epitheliitis: Early-phase fluorescein angiography (left) vs late-phase fluorescein angiography (right). Courtesy of the Clinical Ophthalmology journal, Dove Medical Press.
Diagnostic imaging: indocyanine green angiography
Early and mid-phase indocyanine green angiography shows patchy hyperfluorescence in the macula, and late-phase frames show deep hypocyanescence. These angiographic abnormalities can resolve over time. [115]
 Acute retinal pigment epitheliitis: Indocyanine green angiography: early (left) and late (right) phases. Courtesy of the Clinical Ophthalmology journal, Dove Medical Press.
Acute retinal pigment epitheliitis: Indocyanine green angiography: early (left) and late (right) phases. Courtesy of the Clinical Ophthalmology journal, Dove Medical Press.
Diagnostic imaging: optical coherence tomography scanning
Acute phase spectral domain optical coherence tomography shows disruption of the inner segment-outer segment (IS/OS) junction of the photoreceptors; sometimes a dome-shaped hyper-reflective lesion is seen at the photoreceptor outer segment layer disrupting the ellipsoid zone. [116] The external limiting membrane shows upward displacement, along with RPE undulation and thickening. [117] On disease resolution, the photoreceptor segments, ELM, and RPE band show restoration of normal architecture. Restoration of the foveal IS/OS junction may be of prognostic value for recovery of vision. [118]
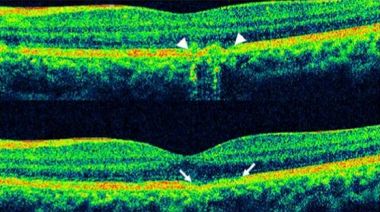 Acute Retinal Pigment Epitheliitis: Disruption of photoreceptor segments, RPE backscattering (top) and RPE undulation (bottom)
Acute Retinal Pigment Epitheliitis: Disruption of photoreceptor segments, RPE backscattering (top) and RPE undulation (bottom)
Electrophysiologic and psychophysical studies
On Amsler grid testing, these patients sometimes describe variable central or paracentral scotomas. [114] Mild color vision abnormalities have also been described by patients with this condition. Results of electroretinography testing are generally normal, whereas electrooculogram testing has yielded various results, from normal to subnormal depending on the extent of RPE involvement. With resolution of the disease, all tests show a high rate of return of normal color vision.
Etiology
Systemic investigations of patients with this rare condition have failed to yield any conclusive etiology, although a viral etiology is generally suspected. Associations with rubella, Borrelia burgdorferi (Lyme disease), influenza, and hepatitis C infections have been reported. [15, 119, 120, 121] In some instances, this condition may be confused with pachychoroidal pigment epitheliopathy. [122] A case of ARPE has been reported following the administration of intravenous bisphosphonate. [123]
Disease course and prognosis
ARPE has a self-limited course, usually with complete resolution over a 6- to 12-week period. [124] Visual acuity and visual field usually return to baseline, and OCT demonstrates restoration of the photoreceptor segment, external limiting membrane and RPE. Clinically, some insignificant residual RPE changes may be noted. Recurrences have been reported but are rare. [125]
Given the generally benign and self-limited course of this condition, no specific treatment is recommended.
Multiple Evanescent White Dot Syndrome
First described in 1984 by Jampol and colleagues, [126, 127] multiple evanescent white dot syndrome (MEWDS) is a self-limited condition that affects young, otherwise-healthy adults, usually in the second to third decades of life, although some cases have been reported in children and elderly patients. [128, 126] The incidence of MEWDS in one 20-year study of white dot syndromes was approximately 0.45 case per 100,000 population per year. [47]
Multiple evanescent white dot syndrome shares many clinical and electrophysiologic features with acute idiopathic blind spot enlargement syndrome (AIBES) and other entities in the acute zonal occult outer retinopathy (AZOOR) group. [4, 7] Despite this commonality, MEWDS is believed to be uniquely distinguishable, warranting its own separate identification. [10, 129]
The condition usually presents unilaterally, with few instances of subsequent involvement of the fellow eye. [130] The condition is primarily seen in women (approximately 75% of patients) and in individuals with significant myopia. [131]
Signs and symptoms
Patients with MEWDS present with a sudden decrease in vision, photophobia, scotomas, an enlarged blind spot, photopsia, and dyschromatopsia. In half of cases, patients report a history of a flulike prodrome in the weeks preceding the onset of ocular symptoms. [132] Initial visual acuity on presentation varies from 20/25 to 20/300 (average 20/100).
The initial manifestation of MEWDS is transient gray-white dots (100-200μm) in the outer retina and retinal pigment epithelium (RPE) with foveal granularity. [126] Additional features may include edematous appearance of the optic nerve and vitreous cells, despite the absence of any obvious anterior segment or external ocular signs of inflammation. [133] It has been reported that eyes with MEWDS have mild aqueous flare on laser flare photometry and lower intraocular pressure than the unaffected fellow eye.
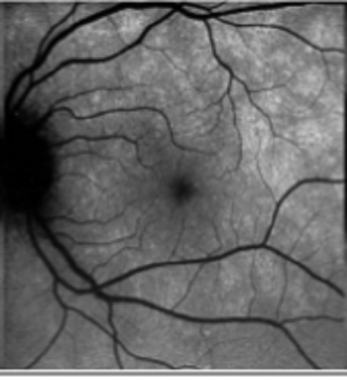
On clinical examination, the early lesions appear to be at the level of the outer retina or RPE and are typically distributed in perifovea, the peripapillary region, and the periphery. Later, the macula often exhibits a fine, granular, white- or orange-speckled appearance. [134, 135] Dots and spots seem to be absent in the foveal area, despite the foveal granularity typically seen on clinical examination.
In the majority of patients with MEWDS, an underlying inflammation of the peripapillary circulation seems to be present, which accounts for the characteristic enlargement of the blind spot. [136]
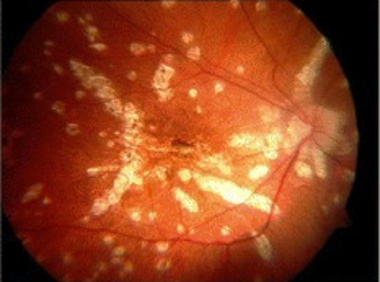
 Fundus photo of the left eye demonstrating optic nerve head edema, foveal granularity, and a circinate distribution of retinal pigment epithelium (RPE) white dots. Courtesy of the Journal of Ophthalmic Inflammation and Infection, BioMed Central Ltd, Springer Nature.
Fundus photo of the left eye demonstrating optic nerve head edema, foveal granularity, and a circinate distribution of retinal pigment epithelium (RPE) white dots. Courtesy of the Journal of Ophthalmic Inflammation and Infection, BioMed Central Ltd, Springer Nature.
 Multiple evanescent white dot syndrome. This patient has spots similar to those seen in the patient in the previous image, but this patient also displays the almost pathognomonic granular changes in the macula.
Multiple evanescent white dot syndrome. This patient has spots similar to those seen in the patient in the previous image, but this patient also displays the almost pathognomonic granular changes in the macula.
Other reported posterior pole findings include splinter hemorrhages, venous sheathing, panuveitis, hyperemia or edema of the optic nerve head, and, rarely, choroidal neovascularization. [137, 138, 139] Choroidal neovascularization can, in fact, be an initial sign or early manifestation of MEWDS. [140, 141]
Diagnostic imaging: fundus autofluorescence
At presentation, fundus autofluorescence shows hyperautofluorescence of the spots, corresponding to the hypocyanescent lesions seen on indocyanine green angiography. [132] The fundus autofluorescence lesions generally appear less numerous than the focal hypofluorescent spots seen on ICG angiography. [142] With disease resolution, the spots usually evolve into areas of decreased autofluorescence or disappear completely. [143, 144]
 MEWDS fundus autofluorescence: Geographic and confluent areas of hyperautofluorescence during the acute phase of disease.
MEWDS fundus autofluorescence: Geographic and confluent areas of hyperautofluorescence during the acute phase of disease.
Diagnostic imaging: fluorescein angiography
Lesions in patients with MEWDS are best described as “wreath-like” punctate areas of early hyperfluorescence. These angiographic lesions are consistent with window defects in the RPE, retinal vascular abnormalities, or a combination of both. The classic “wreath-like” appearance is thought to be a result of inflammation-mediated retinal capillary permeability. Another explanation for the early hyperfluorescence of these lesions on FA may relate to the excitation of microglia that have been implicated in immune-mediated inflammatory processes of the retina. The stellate configuration of microglial cells in the retina resembles the wreath-like lesions seen on fluorescein angiography in MEWDS. Activation of microglia by inflammation, in conjunction with dilation of the deeper retinal circulation, may contribute to the presenting signs of MEWDS. [145]
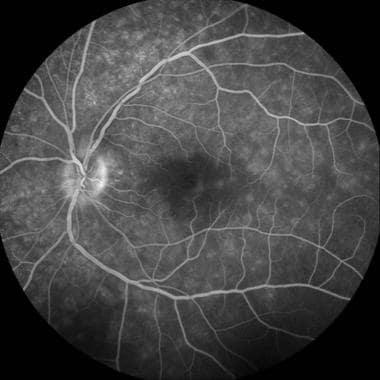 Late-phase fluorescein angiography demonstrating early RPE staining in a parafoveal distribution as well as late leakage around the optic nerve. Courtesy of the Journal of Ophthalmic Inflammation and Infection, BioMed Central Ltd, Springer Nature.
Late-phase fluorescein angiography demonstrating early RPE staining in a parafoveal distribution as well as late leakage around the optic nerve. Courtesy of the Journal of Ophthalmic Inflammation and Infection, BioMed Central Ltd, Springer Nature.
Diagnostic imaging: indocyanine green angiography
Indocyanine green (ICG) angiography demonstrates multifocal hypocyanescent spots and a zonal hypocyanescence in the peripapillary area. Such findings correspond to enlargement of the blind spot on visual field testing. Multiple evanescent white dot syndrome (MEWDS) was traditionally classified as a choriocapillaritis because the typical hypocyanescent lesions visible on ICG angiography were interpreted as hypoperfusion of the innermost choroidal vasculature. [146]
An alternative pathogenesis has been proposed in a multimodal imaging study including optical coherence tomography angiography (OCTA). The authors reported that OCTA images obtained from patients with MEWDS showed no alterations at the level of the choriocapillaris and suggested MEWDS to be a primary photoreceptoritis because pathologic changes are visible on OCT in the outer retina. [147]
Indocyanine green angiography studies have demonstrated multiple deep, small, round, hypofluorescent choroidal lesions appearing early and persisting into the late phases [148] of the disease. Many more lesions are usually observed on ICG angiography than are visible on clinical examination or fluorescein angiography. [149, 150, 151] The hypofluorescent lesions seen on ICG angiography persist longer than lesions displayed during fluorescein angiography. [152, 153] The ICG-defined lesions also closely correlate with visual function. [154] Indocyanine green angiography in MEWDS can be helpful in establishing the diagnosis when fundus findings are subtle and may also provide some useful guidance for therapy. [155, 156]
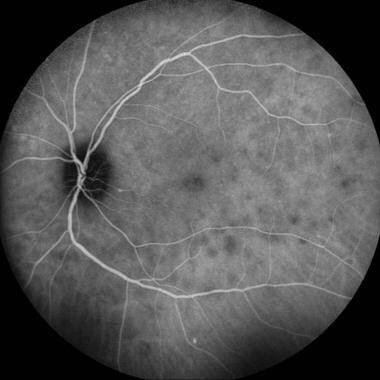 Late-phase indocyanine green angiography demonstrating round hypocyanescent lesions. Courtesy of the Journal of Ophthalmic Inflammation and Infection, BioMed Central Ltd, Springer Nature.
Late-phase indocyanine green angiography demonstrating round hypocyanescent lesions. Courtesy of the Journal of Ophthalmic Inflammation and Infection, BioMed Central Ltd, Springer Nature.
Diagnostic imaging: optical coherence tomography scanning
Ultra-high-resolution optical coherence tomography (OCT) has clearly identified pathologic disruptions of the ellipsoid zone in patients with MEWDS who have no evidence of RPE disturbances or photoreceptor cell body loss. En face OCT imaging demonstrates an unremarkable choroid and choriocapillaris and unaffected RPE layer. The ellipsoid zone and interdigitation zone are disrupted in areas corresponding to spots as well as dots. The en face OCT of the outer portion of the outer nuclear layer shows accumulations of hyperreflective material, which correspond to the dots visible on fluorescein angiography. [157]
At the fovea, OCT shows disruption of the ellipsoid zone and accumulations of hyperreflective material of variable size and shape. These accumulations vary from dome-shaped deposits over the RPE, to linear and vertical aggregations, and other irregular formations centered at the ellipsoid zone. [133]
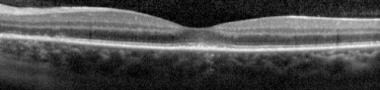 Spectral-domain optical coherence tomography (SD-OCT) demonstrating disruption of the photoreceptor inner segment–outer segment (IS–OS) junction beneath the fovea. Note the intact external limiting membrane. Courtesy of the Journal of Ophthalmic Inflammation and Infection, BioMed Central Ltd, Springer Nature.
Spectral-domain optical coherence tomography (SD-OCT) demonstrating disruption of the photoreceptor inner segment–outer segment (IS–OS) junction beneath the fovea. Note the intact external limiting membrane. Courtesy of the Journal of Ophthalmic Inflammation and Infection, BioMed Central Ltd, Springer Nature.
With resolution of the spots, the OCT scan abnormalities are gradually reversed. [158] Ocular coherence tomography scans obtained through the enlarged blind spot region show thinning of the outer nuclear layer in addition to IS/OS junction defects. [129, 156, 159] Many investigators have concluded based largely on OCT morphology that principal defect in MEWDS occurs at the level of the photoreceptors. [133]
Electrophysiologic and psychophysical studies
Electroretinography performed during the acute phase demonstrates marked reductions in a-wave and early receptor potential (ERP) amplitudes, effectively localizing the disease to the level of the photoreceptors. [127, 160] Selective cone ERG testing demonstrates predominantly more impairment of the S-cone system than of the L- and M-cone systems in the acute stage of MEWDS. [161]
Likewise, multifocal ERG testing has demonstrated reduction of amplitudes not only in areas corresponding to clinically visible lesions but also in the remainder of normal-appearing retina. [162, 163, 164] It has also demonstrated abnormalities in asymptomatic and clinically normal-appearing fellow eyes. [156] Oscillatory potentials are reduced throughout the retina in patients with MEWDS, even in areas with normal multifocal ERG readings. [165] The reduced amplitudes on multifocal ERG testing persist for many months after normalization of both the fundus appearance and the psychophysical test results. [156, 166]
Electrooculography demonstrates only mild abnormalities of the Arden ratio, again suggesting that the abnormalities in patients with MEWDS occur mainly at the level of the photoreceptors and the cycling of visual pigments.
Visual field testing demonstrates not only paracentral and central scotomas but also enlarged blind spots. [129, 167, 168] Similarly, mapping regions of retinal sensitivity using microperimetry during the active phase of the condition demonstrates an enlarged blind spot. [169] Visual evoked potentials (VEPs) have demonstrated prolongation of the P100 latency persisting for many months after resolution of symptoms, suggesting more significant optic nerve involvement than previously thought. [170]
Etiology
The etiology of MEWDS remains unknown. Although the precise pathogenesis is unknown, a virus-like infection with a possible immune-mediated mechanism and genetic susceptibility is suspected. [171] The possibility of a viral trigger has been considered in view of the history of a flulike illness in a significant proportion of patients. A case of an association between recurrence of MEWDS and Herpesviridae family virus infection was reported. In addition, MEWDS has been associated with the following vaccines: hepatitis A and B, yellow fever, human papilloma virus, and meningococcus. [172, 173, 174, 175, 176]
The high prevalence of MEWDS in female patients also suggests that hormonal factors may play a role in this condition. Although findings from systemic workups in patients with MEWDS are usually unremarkable, a weak link between human leukocyte antigen B51 (HLA-B51) and MEWDS has been previously reported, possibly implicating a genetic predisposition for this disease. [177, 178]
As has been suggested, MEWDS might also have an etiology similar to that of multifocal choroiditis. [129, 179] In addition, cases of primary intraocular lymphoma and sympathetic ophthalmia have been described as presenting with an MEWDS-type appearance. [180, 181, 182] This is important in differentiating true etiologic factors from other conditions that mimic MEWDS.
Disease course and prognosis
The disease usually has a self-limited course with good visual recovery, typically occurring within 2 months from the onset of symptoms (varies from 2-16 weeks). Approximately 90% of patients have better than 20/30 final visual acuity. Although late recurrences may occasionally occur, MEWDS is characterized by recovery of visual function and normalization of OCT and ERG findings. [158, 183] In acute MEWDS, the EZ is disrupted but is restored during recovery. No abnormalities are seen in the outer nuclear layer and the RPE during follow-up. Normal peripheral funduscopic appearance returns, although macular changes (ie, foveal granularity) may persist. Long-term follow-up of patients is critical, because in rare cases, choroidal neovascularization can develop. [184, 140] Spaide and colleagues [129] report that AZOOR may develop in patients with MEWDS and that the subsequent development of AZOOR in these patients may be associated with a poorer visual prognosis.
Treatment
Given the self-limited natural history of MEWDS and the low rate of sequelae, no treatment for MEWDS is indicated. Systemic therapy with cyclosporine and corticosteroids has been described for isolated cases, with moderately good results. [185, 186] The use of the anti-vascular endothelial growth factor (anti-VEGF) agent ranibizumab has been effective in treating the rare cases of MEWDS-related macular edema or choroidal neovascularization. [187, 188]
Birdshot (Vitiliginous) Chorioretinopathy
Birdshot chorioretinopathy (BCR) is a chronic, bilateral, posterior uveitis with a distinct clinical phenotype and a strong association with human leukocyte antigen A29 (HLA-A29). Franceschetti and Babel [189] may have incompletely described this chronic condition as early as 1949. First characterized by Ryan and Maumenee [190] in 1980, the condition also has been called vitiliginous chorioretinitis partly because of its fundus appearance and its loose association with cutaneous vitiligo in earlier reports. This disease predominantly affects individuals of middle age (in the fourth decade and beyond) and affects females more often than males. [191]
Signs and symptoms
Early symptoms include floaters, blurred vision, and decreased vision. Later symptoms in the course of the disease include nyctalopia, diminished contrast sensitivity, and decreased color vision. Visual acuity may vary at onset from 20/20 to 20/800, with most patients having a visual acuity of approximately 20/50. Clinically, it presents with subclinical anterior segment inflammation, vitritis, retinal vasculitis of large veins and small retinal capillaries, and rice-shaped hypopigmented choroidal fundus lesions (BCR lesions). [192] The fundus spots may appear even years after the initial presentation of vitritis, vasculitis, and cystoid macular edema.
Specifically, fundus examination reveals vitreous inflammation associated with multiple yellow-white choroiditis spots that are often initially observed inferior to the optic nerve head. These lesions are usually 500-1500 µm in diameter with characteristic nasal, radial distribution in the postequatorial fundus. The typical " BCR" appearance and distribution of lesions gives the disease its name. As the disease progresses, the lesions become more confluent and linear around the veins, eventually becoming more atrophic in appearance.
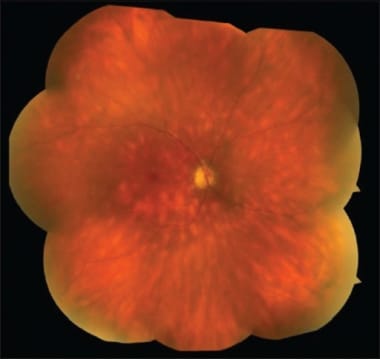 Fundus photograph of BCR. The lesions are at the level of the choroid or retinal pigment epithelium; ovoid, cream-colored with indistinct borders.
Fundus photograph of BCR. The lesions are at the level of the choroid or retinal pigment epithelium; ovoid, cream-colored with indistinct borders.
Although the majority of visual loss in patients with BCR results from cystoid macular edema, other associated ocular findings include [193, 194] :
-
Retinovascular attenuation
-
Retinovascular sheathing
-
Retinal and vitreous hemorrhages
-
Epiretinal membrane formation
-
Glaucoma optic atrophy
-
Rhegmatogenous retinal detachment
-
Subretinal neovascularization
-
Optic disc edema (very rare)
-
Retinal neovascularization (very rare)
-
Serous macular degeneration (very rare)
Diagnostic imaging: fundus autofluorescence
The fundus lesions of BCR generally appear more numerous and are more readily visible with fundus autofluorescence imaging compared with color fundus photographs, especially in eyes with blonde fundi. [195] Fundus autofluorescence shows a variable distribution pattern of lesions: circumferential hypoautofluorescence around the optic nerve head; multifocal hypoautofluorescent areas in the posterior pole, which may or may not correspond to the hypopigmented BCR lesions; and linear hypoautofluorescent streaks along the retinal vessels. [196] Some eyes also show placoid hypoautofluorescence in the macula, indicating retinal pigment epithelium (RPE) atrophy, and this may be an explanation for the decreased visual acuity in patients with BCR. [196]
Diagnostic imaging: fluorescein angiography
The key fluorescein angiographic findings in BCR are leakage of the perifoveal vasculature (cystoid macular edema) and central/peripheral vasculitis. Other angiographic features are inconsistent and vary with age of the lesions and the phase of the study. In the acute phase of BCR, the patches are surprisingly hidden, showing no fluorescein abnormalities. With time and subsequent atrophy of the RPE, the individual lesions behave as window defects transmitting underlying fluorescence from the choroid.
In the early phase of the angiogram, delays in retinal arteriole filling and circulation time are observed, although retinal capillary nonperfusion is not typically seen with this disease. During this phase, the lesions are either not evident or are hypofluorescent. In the late phase, the patches become mildly hyperfluorescent and vasculitis becomes more apparent. In addition, increased retinal circulation time has been observed. [197]
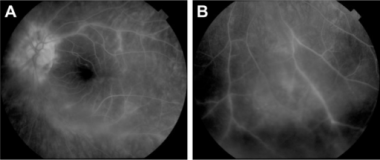 Fluorescein angiogram: Central (A) and peripheral (B) retinal vasculitis associated with birdshot chorioretinopathy. Courtesy of the Clinical Ophthalmology Journal, Dove Press.
Fluorescein angiogram: Central (A) and peripheral (B) retinal vasculitis associated with birdshot chorioretinopathy. Courtesy of the Clinical Ophthalmology Journal, Dove Press.
Diagnostic imaging: indocyanine green angiography
In general, indocyanine green (ICG) angiography can reveal disease activity before it is evident on clinical examination or fluorescein angiography. In addition, the number of lesions is greater than indicated by either fundoscopy or fluorescein angiography. [192] During the active phase of the disease, well-demarcated, hypocyanescent, round-to-oval spots appear throughout the posterior pole with relative sparing of the macula. [58] These spots appear early during the arteriovenous phase and persist unchanged during the late phase of the angiogram.
Progressive diffuse background fluorescence often occurs in the late phase of the angiogram in patients with active disease. In the chronic or burned-out phase of the disease, ICG angiography reveals persistence of the characteristic hypocyanescent spots with absence of diffuse late-phase hyperfluorescence. [198] Remarkably, the ICG lesions are preserved despite an improvement in clinical appearance of the fundus lesions.
Again, ICG angiography may be particularly useful in the early diagnosis of BCR, when the typical BCR lesions are either absent or poorly seen on clinical examination. [199]
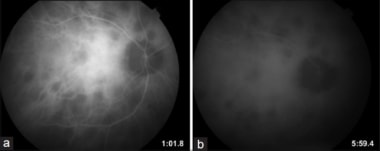 Indocyanine angiography in BCR: Hypo-cyanescent spots in both the early phases of the study (a), as well as the late phases of the study (b) that are more numerous than those seen on fluorescein angiography.
Indocyanine angiography in BCR: Hypo-cyanescent spots in both the early phases of the study (a), as well as the late phases of the study (b) that are more numerous than those seen on fluorescein angiography.
Electrophysiologic and psychophysical studies
An electroretinogram (ERG) obtained from a patient with BCR typically displays moderate-to-severe abnormalities of both rod and cone function (less than or equal to 80% of patients). A disproportionate decrease occurs in the amplitude of the b-wave compared with the a-wave, and some ophthalmologists believe that this decrease is the distinguishing feature of the disease. [200]
BCR retinochoroidopathy electroretinogram response to a scotopic white light stimulus (arrow). Tracing demonstrates a slight disproportionate reduction of the b-wave response in comparison to the a-wave.
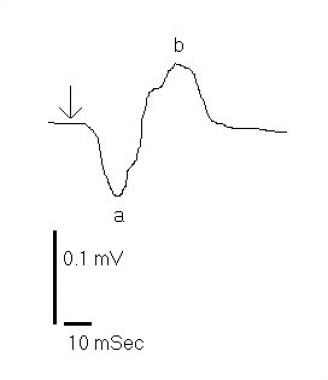 Birdshot retinochoroidopathy electroretinogram response to a scotopic white light stimulus (arrow). Tracing demonstrates a slight disproportionate reduction of the b-wave response in comparison to the a-wave.
Birdshot retinochoroidopathy electroretinogram response to a scotopic white light stimulus (arrow). Tracing demonstrates a slight disproportionate reduction of the b-wave response in comparison to the a-wave.
The 30-Hz flicker implicit time is abnormal in up to 70% of patients with BCR retinochoroidopathy, and normalization of this ERG parameter may be a useful indicator that immunomodulatory therapy can be successfully tapered. [201]
Multifocal ERG (mfERG) has been used to monitor disease progression. Mailac and colleagues [202] demonstrated that progressive deterioration in mfERG during a 5-year period was seen in patients with BCR, whereas classical functional test results were unchanged. Their study suggests a better sensitivity of mfERG in monitoring the retinal function of patients with BCR.
The EOG findings may be either normal or slightly subnormal (70% of patients), and dark adaptation studies may show subnormal rod function. Patients may also exhibit nonspecific color vision defects. The visual field demonstrates a generalized constriction of peripheral fields with central scotomas or enlarged blind spots. These scotomas do not correspond clinically to the BCR lesion distribution. Serial automated visual-field testing has been found to be useful in the monitoring of the response to therapy. [203] Microperimetry of the macula has been suggested as another means of evaluating disease activity and assessing visual impairment in patients with BCR. [204]
Etiology
A genetic predisposition has been suggested because the incidence of the HLA-A29 marker is present in more than 90% of patients with BCR. [205] This incidence rate is to be contrasted with 7% prevalence in the general population. [59] Research has demonstrated that the HLA-A29 gene itself is central to the pathogenesis of the disease. [206] It is thought there may be a genetic mutation in the enzyme ERAP2, which is part of antigen processing for presentation by class I major histocompatibility complex molecules. [206] There is strong evidence of selective antigen processing by ERAP2 with HLA-A29, enabling a unique immunogenic signal leading to BCR. [202]
An autoimmune mechanism as the cause of BCR is supported by several findings. First, in vitro studies demonstrating lymphocyte-mediated immune responses to retinal S-antigen are present in most patients tested. [207, 59] Second, histologic examination reveals a cell-mediated inflammatory response to the outer retina and focal depigmentation of the choroidal melanocytes. [192] BCR also has been associated with cutaneous vitiligo, elevated C4 complement, and elevated soluble interleukin (IL)–2 receptors, [208] as well as elevated intraocular levels of the proinflammatory and T cell–associated cytokines IL-1β, IL-17,and tumor necrosis factor-α. [209]
Therefore, an altered immune mechanism is likely responsible for the clinical findings of this disease. As in the case of multiple evanescent white dot syndrome, primary intraocular lymphoma may masquerade as BCR, and the lack of response to systemic immunosuppression therapy should lead to a reevaluation of the diagnosis. [210]
Disease course and prognosis
Birdshot chorioretinopathy, unlike the aforementioned conditions, is a fairly chronic disease with slow progression. It can be active for 3-4 years before finally going into remission. [207] Although 73% of eyes have a visual acuity better than 20/60, ultimately, one-third of patients have a final visual acuity of less than 20/200. Visual loss in patients with BCR is usually due to refractory cystoid macular edema (CME), macular scarring, development of choroidal neovascular membrane, and cellophane maculopathy. In addition, diffuse retinal dysfunction associated with the long duration of the disease is recognized as a significant risk factor for vision loss.
Treatment
No single therapy has proven effective for treating patients with this condition. Various nonsteroidal anti-inflammatory drugs, corticosteroids, and systemic immunosuppressive agents have been tried with limited efficacy. Corticosteroid therapy in both local and systemic forms often is tried initially for patients with moderate disease. Good control can sometimes be achieved with an initial induction followed by a slow taper to a maintenance dose. [211] In situations in which recurrence and exacerbation of the disease are present, a combination of azathioprine and prednisone may be effective in reducing the relapse rate. [212]
Rush and colleagues [213] reported the use of an intravitreal sustained-release fluocinolone acetonide–containing device in the treatment of 36 eyes of 22 patients with BCR. Their study showed that the fluocinolone intravitreal implant can successfully control intraocular inflammation, decrease cystoid macular edema, and reduce or eliminate the need for systemic immunosuppressive therapy, but at the expense of high incidences of intraocular hypertension, glaucoma, and cataract formation.
Intravenous polyclonal immunoglobulin (IVIG) treatment has been reported to be effective in achieving significant remission without the use of corticosteroids or cytotoxic agents. [214]
Low-dose methotrexate (7.5-25 mg/wk) has been reported to be effective in the treatment of BCR and was associated with better long-term visual outcome compared with no treatment and treatment with corticosteroids. [215]
Cyclosporine A has been demonstrated to have an ameliorating effect on BCR, both as a single agent and in combination with prednisone. [192] A combination of systemic cyclosporine A and mycophenolate mofetil therapy resulted in control of inflammation for at least 1 year in 67% of patients with BCR in 1 study. [216] Studies in which a combination of ketoconazole and cyclosporine were used to treat BCR have shown the combination to be effective not only in controlling the disease, but also achieving stability with much lower doses of cyclosporine than conventionally used. [217, 218]
Biologic agents including infliximab, etanercept, and adalimumab may be considered for use in the treatment of cases of refractory uveitis, although experience with these agents is somewhat limited. [219, 220, 221, 222, 223] Kruh and colleagues [224] reported achieving clinical remission in 72 of 88 patients (81.8%) with refractory noninfectious uveitis, including 13 with BCR, using infliximab.
Serpiginous (Geographic) Choroiditis
Serpiginous choroiditis (SC), is a recurrent, asymmetrically bilateral inflammation of the choroid that leads to loss of choriocapillaris, the overlying retinal pigment epithelium (RPE), of photoreceptor cells. Although the lesions in the eyes of patients with SC are not typically multifocal, many authors include them in the spectrum of white dot syndrome. [225] Serpiginous choroiditis is a relatively rare condition, with a prevalence ranging from 0.2% to 5% of all uveitis patients. [226]
The disease has various aliases in literature, including the following: peripapillary choroiditis, helicoid peripapillary choroidal sclerosis, helicoid peripapillary chorioretinal degeneration, geographic helicoid peripapillary choroidopathy, geographic helicoid choroidopathy, serpiginous choroidopathy, and recently serpiginous-like choroiditis (SLC) [227] . The term serpiginous-like choroiditis describes single or multiple crops of choroiditis lesions progressing in serpentine fashion. In contrast to classical SC, however, the lesions do not start from the optic disc; they are generally multifocal in nature, are associated with an infectious etiology (eg tuberculosis), and affect younger adults. [228]
Serpiginous choroiditis typically affects otherwise healthy patients in the fourth to sixth decades of life. This condition is more prevalent in White individuals, and no predilection for either sex exists.
Signs and symptoms
Patients typically present with decreased central vision, metamorphopsia, or a scotoma. Patients may also remain relatively asymptomatic until the macula is involved. Clinical examination during the acute phase reveals an initial visual acuity of approximately 20/40 (range 20/20 to counting fingers, depending on extent of foveal involvement). In general, no aqueous or vitreous inflammation is seen [229] , but in some studies vitritis has been reported in up to 33% of patients. [230, 231] Although a patient may be initially seen with unilateral disease, all cases of SC eventually become bilateral. The interval between involvement of the primary eye and the contralateral eye may be several years.
The characteristic lesions of SC start as ill-defined grayish-white or creamy yellow patches at the level of deep retina or RPE. [232] The lesion typically arises from the peripapillary region and spreads centrifugally in a geographic or pseudopodlike fashion. The overlying retina may be edematous, and serous retinal detachment may develop in severe cases. The active lesions usually resolve within 6–8 weeks and are characterized by sharpening of the borders with irregular RPE hyperperturbations, diffuse RPE mottling with extensive atrophy of RPE, and choriocapillaris. Sometimes, the atrophy is so extensive that the larger underlying choroidal vessels are exposed because destruction of the entire choroid up to sclera has occurred. Recurrences are common and are usually seen at the edges of previously healed lesions. The interval between recurrent attacks ranges from months to years
Approximately 80% of cases are of the peripapillary variant, and 20% of the macula-involving variant. In some cases the macula may be involved; this variant is termed macular serpiginous choroiditis and, not surprisingly, carries a worse visual prognosis compared with typical serpiginous choroiditis. [233, 234, 235]
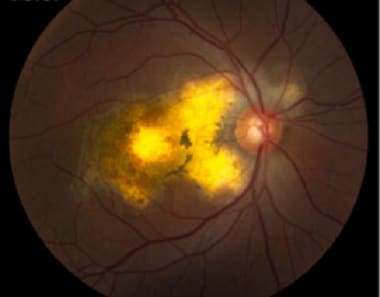 Serpiginous choroiditis (late stage): Color fundus photography showing later stages of disease; geographic RPE atrophy and atrophic lesions surrounding the optic nerve. Courtesy of the Journal of Clinical and Experimental Ophthalmology, Longdom Publishing.
Serpiginous choroiditis (late stage): Color fundus photography showing later stages of disease; geographic RPE atrophy and atrophic lesions surrounding the optic nerve. Courtesy of the Journal of Clinical and Experimental Ophthalmology, Longdom Publishing.
With time, pigment clumping, hypopigmentation of the RPE, and fibrous scar tissue formation occur, and the underlying larger choroidal vessels can be clearly seen within these atrophic patches. [236] Chronic recurrent or untreated disease may result in existing lesions extending to the equator of the eye, and large areas of the fundus may eventually be involved by the disease. Despite bilateral involvement, a high degree of asymmetry between the 2 eyes is often present.
Unfortunately, this condition has a predilection for the temporal rather than the nasal fundus. Although the retina overlying the active lesion is usually edematous, and may develop a serous detachment, the retinal vessels and the optic nerve head usually appear normal throughout the disease. Choroidal neovascularization develops in approximately 25% of patients. [237, 238] Other reported posterior segment complications of serpiginous choroiditis include retinal periphlebitis, retinal and disc neovascularization, branch retinal vein occlusion, RPE detachment, cystoid macular edema, and bilateral macular holes. [239, 240, 241, 242, 243] Tang and colleagues [244] reported a case of antiphospholipid antibody syndrome mimicking the clinical and angiographic appearance of serpiginous choroiditis. Eales disease and acute retinal necrosis have also been reported in association with serpiginous choroiditis. [245, 246, 247]
Diagnostic imaging: fundus autofluorescence
During active disease, serpiginous choroiditis typically presents with large peripapillary hypopigmented helicoid lesions extending outward from the disc in a serpentine configuration. Fundus autofluorescence imaging reveals hypoautofluorescence in areas of atrophy, whereas more active areas are typically hyperautofluorescent.
The identification of new areas of hyperautofluorescence along the borders of old, inactive, hypoautofluorescent lesions is reported to be a sensitive, noninvasive method of detecting disease recurrences. [248, 249] The severity of vision impairment in patients with SC correlates with the degree of foveal hypoautofluorescence. [249]
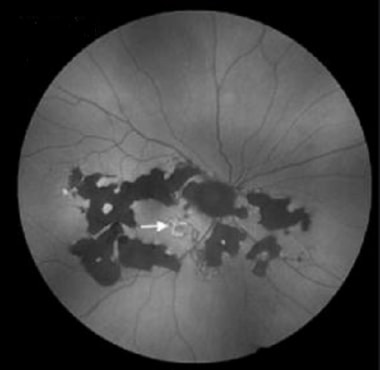 Fundus autofluorescence in SC: Recurrent active disease marked by hyperautofluorescence (white arrow); helical pattern of old inactive lesions (patchy hypoautofluorescence). Courtesy of the Journal of Clinical and Experimental Ophthalmology, Longdom Publishing.
Fundus autofluorescence in SC: Recurrent active disease marked by hyperautofluorescence (white arrow); helical pattern of old inactive lesions (patchy hypoautofluorescence). Courtesy of the Journal of Clinical and Experimental Ophthalmology, Longdom Publishing.
Diagnostic imaging: fluorescein angiography
During the active disease phase, some degree of choroidal hypoperfusion, as well as fluorescence blockage, is caused by edematous RPE and retina. The active lesions are hypofluorescent with fuzzy, irregular borders in early phase of the angiogram. This is followed by leakage of the dye from the choriocapillaris at the borders of the inflamed lesions, which is manifested by gradual hyperfluorescence at the borders of the lesions in the mid-phase of the angiogram. [246]
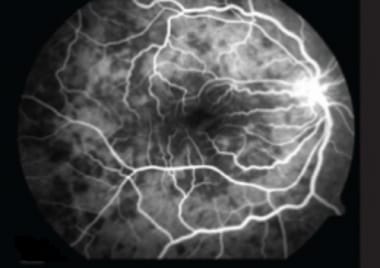 Fluorescein angiogram SC (active stage) showing presence of confluent hypofluorescent lesions radiating away from the disc not involving the fovea with staining around these lesions
Fluorescein angiogram SC (active stage) showing presence of confluent hypofluorescent lesions radiating away from the disc not involving the fovea with staining around these lesions
The inactive stage of the disease is characterized by a patchy diminution of the early choroidal flush resulting from the destruction of the choriocapillaris, and in some cases, of the larger choroidal vessels as well. Unmasking of the underlying choroidal vessel hyperfluorescence in areas of healed lesions, which have become atrophic scars, can be observed. The staining at the margins of inactive lesions results from leakage of adjacent preserved choriocapillaris.
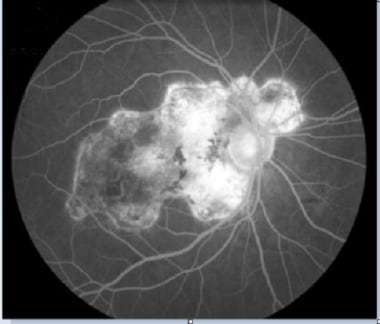 Fluorescein angiogram SC (late stage): Note the late hyperfluorescence (staining) of damaged RPE areas. Courtesy of the Journal of Clinical and Experimental Ophthalmology, Longdom Publishing.
Fluorescein angiogram SC (late stage): Note the late hyperfluorescence (staining) of damaged RPE areas. Courtesy of the Journal of Clinical and Experimental Ophthalmology, Longdom Publishing.
Diagnostic imaging: indocyanine green angiography
Active lesions show blockage of the dye beginning from the early to the late phase on indocyanine green angiography, a feature which is thought to be contributed by a combination of both abnormalities in choroidal perfusion and blockage of fluorescence by the inflamed RPE and outer retina. [250] The extent of involvement of choroidal inflammation observed on ICG angiography is beyond the limits delineated by corresponding fluorescein studies or clinical examination. [251] Similar observations were reported in patients with subacute and healed lesions of SC, for whom ICG angiography can show better and earlier delineation of resolution of choroiditis than corresponding fluorescein changes. [251] Indocyanine green angiography is very helpful in differentiating choroidal neovascularization (CNV) in the presence of active choroiditis because both CNV and choroiditis leak fluorescein. Choroiditis usually shows early hypofluorescence on ICG angiography, whereas hyperfluorescence is observed in CNV.
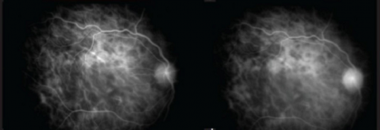 Indocyanine green angiography: Early phase (left) ICGA showing presence of numerous confluent hypocyanescent lesions around the disc and surrounding the fovea; Late phase (right) Persistence of the hypocyanescent lesions with involvement of fovea with areas of loss of choriocapillaris on ICGA
Indocyanine green angiography: Early phase (left) ICGA showing presence of numerous confluent hypocyanescent lesions around the disc and surrounding the fovea; Late phase (right) Persistence of the hypocyanescent lesions with involvement of fovea with areas of loss of choriocapillaris on ICGA
Diagnostic imaging: optical coherence tomography
The active SC lesion typically demonstrates hyperreflectivity of the outer retina with loss of inner and outer segment (IS/OS) junction and loss of RPE in subsequent phases of inflammation. Extensive mononuclear cell infiltrate in the choriocapillaris and choroid, as reported in histopathology, can be observed as early choroidal hyperreflectivity, which is often described as a “waterfall” effect. [252, 253] Spectral-domain OCT typically shows hyperreflectivity of both the outer retina and choroid along with disruption of the IS/OS junction. [254, 255]
Ocular coherence tomography angiography (ACT-A) in patients with SC is a diagnostic modality that obtains high-resolution images of retinal and choroidal vasculature by detecting intravascular blood flow based on split-spectrum amplitude-decorrelation angiography without injecting the dye. OCT-A in serpiginous choroiditis demonstrated decreased vascularity on choriocapillaris but intact retinal vascularity. [256]
Electrophysiologic and psychophysical studies
The degree of abnormality detected by electroretinography (ERG) and electrooculography (EOG) correlates closely with the extent of clinical involvement of the retina. [239] Unless retinal involvement is extensive, as in long-standing or untreated disease, the findings from ERG and EOG studies are usually normal or only mildly subnormal. [257] Visual-field testing reveals absolute or relative scotomas corresponding to clinical retinal lesions. Central, cecocentral, and paracentral scotomas are the predominant feature on visual-field testing.
Microperimetry has revealed dense scotomas corresponding to all atrophic or inactive lesions and some active lesions, and relative scotomas in clinically normal–appearing areas of the posterior pole and peripapillary areas, indicating possible subclinical inflammation or choroidal perfusion abnormalities. [258] In spite of extensive involvement of the macula in serpiginous choroiditis, stable fixation was observed in more than 60% of eyes by microperimetry. [258]
Etiology
The etiology of SC remains elusive, although it is known that the lesions appear to be at the level of the RPE and choriocapillaris. Histopathologic studies reveal that the disease is primarily a nongranulomatous choroiditis with a moderate, diffuse lymphocytic infiltration of the choroid. [259] The suggestion that SC may be caused by immunologic mechanisms is strengthened by its association with the human leukocyte antigen A2 (HLA-A2) and human leukocyte antigen B7 (HLA-B7) histocompatibility and by the occurrence of other inflammatory lesions in the involved eye. [240] Evidence of serologic reaction to retinal S-antigen in affected patients lends further support to alterations in immune responsiveness. [260] Other associations with serpiginous and serpiginous-like choroiditis include sarcoidosis, tuberculosis, extrapyramidal dystonia, and elevated factor VIII-von Willebrand factor. [239, 261, 262, 263]
Disease course and prognosis
The disease is characterized by an episodic, but generally indolent and progressive, course with recurrences often going undetected until the macula is involved. At intervals varying from weeks to years, the patient is subject to recurring episodes of activity that each time involve a new and usually contiguous area of the fundus. [21] Active lesions typically resolve in 6-8 weeks, but reactivations may not occur for months or even years. Fibrous metaplasia of the RPE develops within the area of chorioretinal atrophy in approximately 50% of patients. This is particularly prominent in the nasal and peripapillary regions.
Serpiginous lesions in the macula, cystoid macular edema, and choroidal neovascularization (CNV) can adversely affect central visual acuity. Choroidal neovascularization is the most common complication associated with SC. The reported incidence of CNV in patients with SC ranges from 10 to 25%. [264] CNV typically arises close to the edge of choroidal lesions and can occur in both active and healed choroid. The contralateral eye may be involved within months or several years following the initial process in the involved eye. Christmas and colleagues [265] found that 25% of patients have a visual acuity of 20/200 or worse in one eye on long-term follow-up. Most patients, however, can maintain good central and peripheral function in at least 1 eye.
Treatment
Treatment typically consists of a combination of the following agents:
- Corticosteroids - Management of macula-threatening SC usually involves combination treatment with high-dose corticosteroids (oral prednisone, 1 mg/kg/day or 1 g of pulse intravenous methylprednisolone for 3-5 days, followed by a tapering dose of oral prednisone over 1-3 months) to control the acute inflammatory disease and an immunosuppressive agent or immunomodulator provided to prolong remission and preserve good vision. [266, 267, 268, 269]
- Immunosuppressive Agents - Agents such as methotrexate, azathioprine, cyclosporine, chlorambucil, or cyclophosphamide can help to attain longer period of disease inactivity and reduce the risk of potential adverse effects associated with high-dose systemic steroids. [227] A triple-agent immunosuppressive regimen consisting of cyclosporine (5 mg/kg/day initially), azathioprine (1.5 mg/kg/day), and prednisolone (1 mg/kg/day) was found to be effective. [270]
- Intravitreal Agents - Intravitreal steroids have been used, as has the fluoroquinolone acetonide intravitreal implant as alternatives to long-term systemic steroid use with its attendant complications. [271, 272, 273, 274, 275]
- Biologics (eg, infliximab) - have been shown to be useful in the management of many uveitic conditions, including SC, refractory to other modalities of treatment. Many authors have recommended the use of biologic therapy for patients with recalcitrant cases of SC for whom other modalities of treatment have failed. [276]
- Intravitreal anti-VEGF agents - Bevacizumab and ranibizumab have been used to successfully manage the complication of choroidal neovascularization in patients with SC. [277, 278, 279, 280, 281]
Diffuse Unilateral Subacute Neuroretinitis
Diffuse unilateral subacute neuroretinitis (DUSN), which is caused by the chronic subretinal migration of 1 of at least 2 species of nematodes, characteristically effects children or young adults with a slight male preponderance. Patients are otherwise healthy and are often asymptomatic at the time of presentation. [282] Diffuse unilateral subacute neutoretinitis is caused by the migration of worm into the ocular tissues. The entry of the worm into the eye occurs when nematode eggs, after being ingested, find their way into the eye via hematogenous spread from the gastrointestinal tract. For months to years, the worm can wander in the subretinal space, eventually causing inflammation in the tissues. [283]
Signs and symptoms
Patients often present quite late in course of the disease with typically poor visual acuity, usually less than 20/200. Intraocular inflammation tends to be unilateral and diffuse, and in the early stages may present as vision loss, vitritis, papillitis and clusters of multiple evanescent, fleeting gray-white outer retinal lesions. In later disease stages, optic atrophy, retinal arteriolar narrowing, and diffuse retinal pigment epithelium (RPE) changes can be seen. [284]
The characteristic posterior pole findings are focal, gray-white or yellow-white spots developing in the deep layers of the retina and also at the level of the RPE. These normally develop within several days to weeks from the onset of symptoms. The lesions tend to cluster in the macula or juxtamacular region and typically last several days before fading. Occasionally, a subretinal worm (500-2000 µm long, 25 µm wide) can be seen.
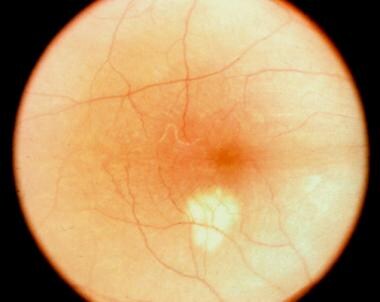 Diffuse unilateral subacute neuroretinitis. Attempted focal laser photocoagulation of a subretinal nematode suspected of being Baylisascaris procyonis. Courtesy of Everett Ai, MD, San Francisco, Calif.
Diffuse unilateral subacute neuroretinitis. Attempted focal laser photocoagulation of a subretinal nematode suspected of being Baylisascaris procyonis. Courtesy of Everett Ai, MD, San Francisco, Calif.
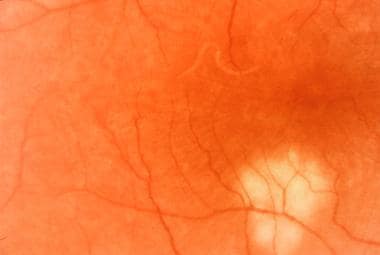 The worm was only later discovered to have migrated from the original site, which initially was targeted for laser treatment.
The worm was only later discovered to have migrated from the original site, which initially was targeted for laser treatment.
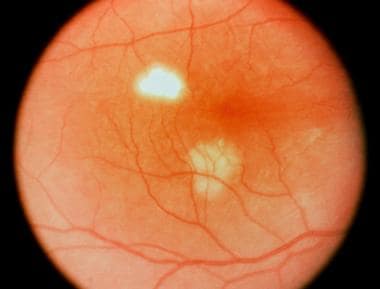
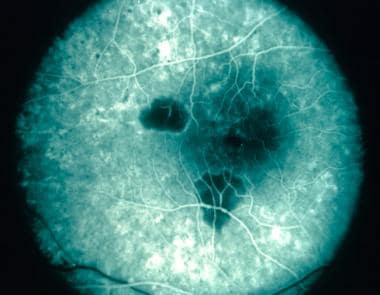 The same eye immediately after the second laser treatment, showing not only the hypofluorescent laser-treated areas but also the characteristic mottled hyperfluorescence of diffuse retinal pigment epithelium and choriocapillaris destruction.
The same eye immediately after the second laser treatment, showing not only the hypofluorescent laser-treated areas but also the characteristic mottled hyperfluorescence of diffuse retinal pigment epithelium and choriocapillaris destruction.
In the inactive phase, the posterior pole exhibits extensive mottling of the RPE, assuming a pseudo–retinitis pigmentosa appearance. Extensive retinovascular narrowing and sheathing with optic atrophy are also seen. Subretinal neovascularization may develop at this stage. A mild to moderate anterior chamber and vitreous cell reaction may be present even in the inactive stage. The patient often has an afferent pupillary defect. Barbazetto and colleagues [285] reported that the active phase of DUSN can mimic the fundus appearance of a white dot syndrome.
Diagnostic imaging: fluorescein angiography
In the very early stage of the disease, angiographic findings may be entirely normal. If lesions are present, hypofluorescence of the lesions occurs during the early phase of the angiogram, and the lesions typically stain during the late phase. In addition, leakage occurs from the capillaries overlying the optic nerve, in addition to more diffuse perivenous leakage occurring in the retina. In a more typical late stage of the disease, an increase in the background choroidal fluorescence occurs as a result of to extensive RPE dropout and loss of pigmentation. Hyperfluorescence, indicative of RPE atrophy, occurs in the periphery and the peripapillary regions, exhibiting fine, mottled hyperfluorescence, particularly in the macular region. If a worm is present, it can be seen as a hypofluorescent lesion.
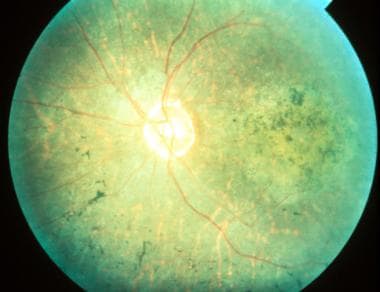 Diffuse unilateral subacute neuroretinitis. Late stage of the disease presenting the characteristic pseudo–retinitis pigmentosa features.
Diffuse unilateral subacute neuroretinitis. Late stage of the disease presenting the characteristic pseudo–retinitis pigmentosa features.
Electrophysiologic and psychophysical studies
Often, a moderate-to-severe reduction in the ERG occurs. With later stages of the disease, a disproportionate decrease occurs with the b-wave amplitude relative to the decrease in the a-wave amplitude. In rare instances, the ERG may be extinguished.
Etiology
Leading the list of suspected causative agents in this disease are the nematodes Toxocara canis (dog roundworm), Ancylostoma caninum (dog hookworm), and Baylisascaris procyonis (raccoon intestinal nematode). [286, 287] Clinically, 2 types of worms have been described in association with this condition. They are differentiated by length. Smaller worms (400-1000 µm) and longer worms (1500-2000 µm) may wander in the subretinal space for many years and cause progressive ocular damage. However, the nematode may be found during any stage of the disease.
The worm is often located in deep, white retinal exudative lesions. Surgical removal of a subretinal nematode in a patient with DUSN identified the causative organism as a third-stage T canis larva. [288] The result of serologic testing for Toxocara is typically negative in patients with DUSN.
The damage caused by the organisms appears to involve a local toxic tissue reaction to the outer retina caused by the worm's byproducts, in addition to a more diffuse toxic reaction affecting both the inner and outer retinal tissues, which is believed to be due to the host's immunologic defenses. Eosinophilia is detected infrequently, and patients do not manifest evidence of systemic disease.
Histopathologic examination of the few eyes with DUSN that have been examined reveals a largely nongranulomatous vitritis, retinitis, and retinal and optic nerve perivasculitis. Low-grade, patchy, nongranulomatous choroiditis is also present. Extensive degeneration of the peripheral retina occurs, particularly of the posterior region within the arcades. Mild-to-moderate degenerative changes in the RPE have also been described. Not surprisingly, optic atrophy is a common feature of these eyes.
Disease course and prognosis
After the acute phase of the disease, focal and more diffuse depigmentation of the RPE develops over the ensuing weeks or months. Pigment migration occurs into the overlying retina with a gradual narrowing of the retinal vasculature. Retinal hemorrhages may result from choroidal neovascularization and can result in occasional serous retinal detachments.
Treatment
Treatment of patients with DUSN has not been optimized. Argon laser photocoagulation is effective in destroying the worm but can cause an inflammatory reaction. Surgical (transvitreal) removal of a subretinal nematode elicits less inflammatory response but is accompanied by higher risks inherent to this treatment. Antihelminthics (eg, thiabendazole, diethylcarbamazine) or antifilarial agents (eg, ivermectin) may be used as adjunctive treatment. Oral antihelminthics in conjunction with laser therapy have been employed in most reported cases, either as a single dose or with treatment courses up to 3 weeks. [289]
Conclusion
Conclusion
With the exception of DUSN, the idiopathic multifocal choroidopathy syndromes discussed remain problematic, not only from an etiologic point of view but also from a classification point of view. Although there is a practical benefit to combine these entities under one group, such as acute zonal occult outer retinopathy or multifocal choroidopathy, sufficiently unique characteristics in each of these conditions still exist to make them distinct from each other. Additionally, many instances occur in which the patient's symptomatology and clinical findings overlap between the entities. Similarly, although many findings point to an infectious and/or immunologic mechanism as the cause of these conditions, no conclusive evidence for either has yet been demonstrated. Until more clinical and histologic information regarding these entities is available, attempts at grouping or subgrouping these conditions will be inadequate in the long run.
-
Multifocal choroiditis and panuveitis. Multiple, discrete lesions appearing in the early stage of the disease.
-
Multifocal choroiditis and panuveitis. Late stage of disease manifesting atrophic, punched-out lesions.
-
Diffuse subretinal fibrosis syndrome. This patient's right eye shows typical subretinal angulated scar formation.
-
Diffuse subretinal fibrosis syndrome. Midphase of fluorescein angiogram demonstrating progressive leakage hyperfluorescence.
-
Diffuse subretinal fibrosis syndrome. Late phase of the fluorescein angiogram demonstrating progressive leakage as hyperfluorescence.
-
Multiple evanescent white dot syndrome. This patient has spots similar to those seen in the patient in the previous image, but this patient also displays the almost pathognomonic granular changes in the macula.
-
Multiple evanescent white dot syndrome. Fluorescein angiogram of the same patient as in the image directly previous to this one, demonstrating the characteristic late phase mottled hyperfluorescence and staining of the optic disc.
-
Midphase of fluorescein angiogram of the same patient as in the image directly above, demonstrating typical light leakage and staining of the lesions.
-
Acute retinal pigment epitheliitis.
-
Serpiginous (geographic) choroiditis. This patient shows an old, inactive serpiginous scar extending in a pseudopod-like fashion from the optic disc. Courtesy of Patrick Monahan, MD, San Jose, Calif.
-
Serpiginous (geographic) choroiditis. Chronic atrophic lesions that display widespread geographic chorioretinal atrophy with moderate foveal sparing.
-
Serpiginous (geographic) choroiditis. A transit-phase fluorescein angiogram of the same eye as in the above image demonstrates leakage and staining hyperfluorescence at the margins of the atrophic lesions. The underlying large choroidal vessels are seen easily within the atrophic lesions.
-
Birdshot (vitiliginous) retinochoroidopathy. Multiple, uniformly hypopigmented lesions with mild disc swelling.
-
Birdshot (vitiliginous) retinochoroidopathy. Late phase fluorescein angiogram that demonstrates patchy hyperfluorescence.
-
Birdshot retinochoroidopathy electroretinogram response to a scotopic white light stimulus (arrow). Tracing demonstrates a slight disproportionate reduction of the b-wave response in comparison to the a-wave.
-
Diffuse unilateral subacute neuroretinitis. Attempted focal laser photocoagulation of a subretinal nematode suspected of being Baylisascaris procyonis. Courtesy of Everett Ai, MD, San Francisco, Calif.
-
The worm was only later discovered to have migrated from the original site, which initially was targeted for laser treatment.
-
A second laser treatment was successful at destroying the worm.
-
The same eye immediately after the second laser treatment, showing not only the hypofluorescent laser-treated areas but also the characteristic mottled hyperfluorescence of diffuse retinal pigment epithelium and choriocapillaris destruction.
-
Diffuse unilateral subacute neuroretinitis. Late stage of the disease presenting the characteristic pseudo–retinitis pigmentosa features.
-
Fluorescein angiography in APMPPE: Early vs late phases. Courtesy of the Journal of Clinical and Experimental Ophthalmology, Longdom Publishing.
-
Note the disruption of RPE/outer retina that corresponds to placoid lesions (white arrows). Courtesy of the Journal of Clinical and Experimental Ophthalmology, Longdom Publishing.
-
Acute retinal pigment epitheliitis: Early-phase fluorescein angiography (left) vs late-phase fluorescein angiography (right). Courtesy of the Clinical Ophthalmology journal, Dove Medical Press.
-
Acute retinal pigment epitheliitis: Indocyanine green angiography: early (left) and late (right) phases. Courtesy of the Clinical Ophthalmology journal, Dove Medical Press.
-
Late-phase fluorescein angiography demonstrating early RPE staining in a parafoveal distribution as well as late leakage around the optic nerve. Courtesy of the Journal of Ophthalmic Inflammation and Infection, BioMed Central Ltd, Springer Nature.
-
Fundus photo of the left eye demonstrating optic nerve head edema, foveal granularity, and a circinate distribution of retinal pigment epithelium (RPE) white dots. Courtesy of the Journal of Ophthalmic Inflammation and Infection, BioMed Central Ltd, Springer Nature.
-
Late-phase indocyanine green angiography demonstrating round hypocyanescent lesions. Courtesy of the Journal of Ophthalmic Inflammation and Infection, BioMed Central Ltd, Springer Nature.
-
Spectral-domain optical coherence tomography (SD-OCT) demonstrating disruption of the photoreceptor inner segment–outer segment (IS–OS) junction beneath the fovea. Note the intact external limiting membrane. Courtesy of the Journal of Ophthalmic Inflammation and Infection, BioMed Central Ltd, Springer Nature.
-
Fluorescein angiogram: Central (A) and peripheral (B) retinal vasculitis associated with birdshot chorioretinopathy. Courtesy of the Clinical Ophthalmology Journal, Dove Press.
-
Serpiginous choroiditis (late stage): Color fundus photography showing later stages of disease; geographic RPE atrophy and atrophic lesions surrounding the optic nerve. Courtesy of the Journal of Clinical and Experimental Ophthalmology, Longdom Publishing.
-
Fundus autofluorescence in SC: Recurrent active disease marked by hyperautofluorescence (white arrow); helical pattern of old inactive lesions (patchy hypoautofluorescence). Courtesy of the Journal of Clinical and Experimental Ophthalmology, Longdom Publishing.
-
Fluorescein angiogram SC (late stage): Note the late hyperfluorescence (staining) of damaged RPE areas. Courtesy of the Journal of Clinical and Experimental Ophthalmology, Longdom Publishing.
-
Multifocal choroiditis: Late phase of fluorescein angiogram showing staining of inactive lesions. Courtesy of the Journal of Clinical and Experimental Ophthalmology, Longdom Publishing.
-
Color fundus photography images show multiple bilateral yellow-white placoid lesions, at the level of external retina, posterior to the equator, and a slight fade of the edges of the optic nerve. Courtesy of the Case Reports in Ophthalmological Medicine journal, Hindawi.
-
Fundus autofluorescence: Hypoautoflourescence corresponding to the placoid lesions, with hyperautofluorescent edges. Courtesy of the Case Reports in Ophthalmological Medicine journal, Hindawi.
-
APMPPE Indocyanine green angiography: Hypofluorescence throughout all phases of the angiogram, corresponding to the placoid lesions. Courtesy of the Case Reports in Ophthalmological Medicine journal, Hindawi.
-
SD-OCT: Note the hyperreflectivity from the outer plexiform layer to the RPE. The retina is of normal thickness. Courtesy of the Case Reports in Ophthalmological Medicine journal, Hindawi.
-
Late stages of multifocal choroiditis: Note the multiple atrophic lesions. Courtesy of the Case Reports in Ophthalmological Medicine journal, Hindawi.
-
Multifocal choroiditis and panuveitis (early stage): Multiple, white, discrete lesions at the level of the choroid. Courtesy of the Journal of Ophthalmic Inflammation and Infection, BioMed Central Ltd, Springer Nature.
-
Early disease stage (top): Fluorescein angiography reveals discrete, roundish hypofluorescent lesions, which are clinically unapparent, but correspond to future lesions. Late disease stage (bottom): Hypofluorescent lesions have considerably increased in number and are hyperfluorescent in late phase of the fluorescein angiography. A discrete macular late leakage is found in the right eye consistent with choroidal neovascularization. Courtesy of the Journal of Medical Case Reports, BioMed Central Ltd, Springer Nature.
-
Serpigionous OCT: Outer retinal loss and RPE disruption in the areas of active lesions. Courtesy of the Journal of Clinical and Experimental Ophthalmology, Longdom Publishing.
-
OCT hallmarks of BCR: a) Disruption of the photoreceptor junction; b) distinct hyperechogenic line and diffuse retinal thinning; c) well-demarcated transition zone between affected and un-affected retina. Courtesy of the Orphanet Journal of Rare Diseases, BioMed Central Ltd, Springer Nature.
-
Early phase ICG in retinochoroidopathy: Note the hypocyanescent lesions. Courtesy of the Journal of Ophthalmic Inflammation and Infection, BioMed Central Ltd, Springer Nature.
-
Late-phase ICG in birdshot retinochoroidopathy: Hypocyanescent lesions are more prominent, as is diffuse background cyanescence of the posterior pole. Courtesy of the Journal of Clinical and Experimental Ophthalmology, Longdom Publishing.
-
Early phase ICG (top) and Late phase ICG (bottom): The lesions remained hypocyanescent throughout all phases of the angiogram
-
Fluorescein angiogram in a patient with APMPPE: Early (left) and intermediate (right) phases show hyperfluorescence with moderate staining of lesions.
-
Early phase (left) and late phase (right) frames reveal more extensive areas of involvement (hypofluorescence) than seen on clinical examination.
-
OCT of APMPPE Lesions: Note hyper-reflectivity at the level of the RPE.
-
Acute Retinal Pigment Epitheliitis: Disruption of photoreceptor segments, RPE backscattering (top) and RPE undulation (bottom)
-
MEWDS fundus autofluorescence: Geographic and confluent areas of hyperautofluorescence during the acute phase of disease.
-
Fundus photograph of BCR. The lesions are at the level of the choroid or retinal pigment epithelium; ovoid, cream-colored with indistinct borders.
-
Indocyanine angiography in BCR: Hypo-cyanescent spots in both the early phases of the study (a), as well as the late phases of the study (b) that are more numerous than those seen on fluorescein angiography.
-
Serpiginous Choroiditis (active phase): Color fundus photograph showing presence of yellowish white subretinal confluent lesions radiating away from the disc sparing the fovea.
-
Fluorescein angiogram SC (active stage) showing presence of confluent hypofluorescent lesions radiating away from the disc not involving the fovea with staining around these lesions
-
Indocyanine green angiography: Early phase (left) ICGA showing presence of numerous confluent hypocyanescent lesions around the disc and surrounding the fovea; Late phase (right) Persistence of the hypocyanescent lesions with involvement of fovea with areas of loss of choriocapillaris on ICGA
Tables

What would you like to print?
- Overview
- Multifocal Choroiditis
- Acute Posterior Multifocal Placoid Pigment Epitheliopathy
- Acute Retinal Pigment Epitheliitis (Krill Disease)
- Multiple Evanescent White Dot Syndrome
- Birdshot (Vitiliginous) Chorioretinopathy
- Serpiginous (Geographic) Choroiditis
- Diffuse Unilateral Subacute Neuroretinitis
- Conclusion
- Show All
- Media Gallery
- References



