Approach to the Uveitis Workup
For many years, uveitis was considered a single disease entity; therefore, the approach to treatment varied very little. As knowledge of the disease process grew and the sophistication of immunologic and microbiologic testing increased, the fact that uveitis entails a multitude of diseases became clear. Although some diseases are local ocular immune phenomena, many of them are systemic diseases with ocular manifestations. Because the spectrum of disease pathogenesis ranges from autoimmunity to neoplasia to viruses, the practitioner of uveitis requires an understanding of internal medicine, infectious diseases, rheumatology, and immunology.
Patients with uveitis can present with some of the most challenging diagnostic dilemmas in all of ophthalmology. Because the treatment and prognosis of various uveitic entities varies greatly, accurate diagnosis is imperative. Many diseases, including Fuchs uveitis syndrome (formerly known as Fuchs heterochromic iridocyclitis), Behçet disease, toxoplasmosis, cytomegalovirus (CMV) retinitis, ocular histoplasmosis, and Vogt-Koyanagi-Harada (VKH) disease, are clinical diagnoses that require little, if any, laboratory analysis. Likewise, the patient presenting with an initial episode of acute nongranulomatous anterior uveitis and an unremarkable review of systems and physical examination does not require a laboratory evaluation.
Laboratory tests are rarely useful as screening tools for this disease. In determining which tests to order, using clues from the history and physical examination and knowledge of the pretest probability of the disease in question is helpful. This diagnostic process is important to avoid false-positive results and costly and unnecessary testing. Consequently, no standard laboratory evaluation exists for the patient with uveitis, except in screening for syphilis and possibly sarcoidosis, both of which can present in a myriad of ways. The key to the targeted and efficient patient evaluation is a thorough history, physical examination, and review of systems. With this information, the practitioner can generate a differential diagnosis and a subsequent strategy for laboratory evaluation.
The lists below include some of the most common systemic findings in patients with uveitis.
Head/CNS
Evaluation of the head and CNS includes the following:
-
Headaches - VKH disease, sarcoidosis, Behçet disease, tuberculosis, herpes zoster, large cell lymphoma, polyarteritis nodosa (PAN), Cryptococcus meningitis, toxoplasmosis, Lyme disease
-
Auditory/vestibular - VKH disease, sarcoidosis, Wegener granulomatosis, Eales disease, syphilis
-
Cranial neuropathy - Lyme disease, sarcoidosis, multiple sclerosis, syphilis, herpes simplex virus
-
Psychosis - VKH disease, Behçet disease, systemic lupus erythematosus (SLE)
-
Cerebral vasculitis - Acute posterior multifocal placoid pigment epitheliopathy (APMPPE), Behçet disease, herpes simplex virus, herpes zoster virus, syphilis, Lyme disease
Ear/Nose/Throat
Evaluation of the ears, nose, and throat include the following:
-
Bilateral ear pinna inflammation - Relapsing polychondritis
-
Saddle nose deformity - Syphilis, Wegener granulomatosis, relapsing polychondritis
-
Oral ulcers - Behçet disease, SLE, herpes simplex, Reiter syndrome, ulcerative colitis
-
Sinusitis - Sarcoidosis, Wegener granulomatosis
-
Salivary/lacrimal gland swelling - Sarcoidosis, lymphoma
-
Lymphadenopathy - Lymphoma, HIV, toxoplasmosis
Gastrointestinal
Evaluation of the gastrointestinal system includes the following:
-
Diarrhea -Crohn disease, ulcerative colitis, Whipple disease
-
Jaundice/hepatosplenomegaly -Brucellosis, CMV, sarcoidosis, infectious hepatitis, autoimmune hepatitis
Pulmonary
Evaluation of the pulmonary system includes the following:
-
Cough, shortness of breath - Tuberculosis, sarcoidosis, Pneumocystis carinii, malignancy, Wegener granulomatosis
-
Nodules, hilar adenopathy, infiltrates - Ocular histoplasmosis, sarcoidosis (hilar adenopathy), malignancy, tuberculosis, P carinii pneumonia
Genitourinary
Evaluation of the genitourinary system includes the following:
-
Genital ulcers - Behçet disease, Reiter syndrome, syphilis
-
Hematuria - Wegener granulomatosis, PAN, SLE
-
Circinate balanitis -Ankylosing spondylitis, Reiter syndrome
-
Urethral discharge - Reiter syndrome, syphilis
-
Nephritis - PAN, Wegener granulomatosis, tubulointerstitial nephritis and uveitis (TINU)
-
Epididymitis - PAN, Behçet disease, Reiter syndrome
Dermatologic
Evaluation of the dermatologic system includes the following:
-
Alopecia - VKH disease, syphilis
-
Vitiligo, poliosis - VKH disease
-
Nodules - Sarcoidosis, SLE, leprosy, Crohn disease, ulcerative colitis
-
Rash - Syphilis, Lyme disease, Reiter syndrome, leprosy, sarcoidosis, herpes zoster, Behçet disease, psoriasis, SLE, Kawasaki disease
-
Keratoderma blennorrhagicum - Reiter syndrome, ankylosing spondylitis
-
Erythema nodosum - Behçet disease, sarcoidosis, APMPPE
Musculoskeletal
Evaluation of the musculoskeletal system includes the following:
-
Arthralgias/arthritis - Behçet disease, sarcoidosis, SLE, juvenile idiopathic arthritis (JIA), Lyme disease, syphilis, psoriatic arthritis, Reiter syndrome, ulcerative colitis
-
Sacroiliitis - Ankylosing spondylitis, Reiter syndrome, inflammatory bowel disease
Constitutional
Constitutional evaluation includes the following:
-
Fever - Reiter syndrome, Behçet disease, PAN, inflammatory bowel disease, HIV, tuberculosis, coccidioidomycosis, Whipple disease
-
Night sweats - Malignancy, tuberculosis, sarcoidosis, coccidioidomycosis
-
Flulike symptoms - APMPPE, multiple evanescent white dot syndrome (MEWDS)
Opremcak advocates a differential-based diagnostic system that primarily uses three clinical features, as follows: (1) location of the inflammation in the eye (corneoscleral, anterior, intermediate, posterior, or diffuse uveitis), (2) type of inflammation (granulomatous vs nongranulomatous), and (3) associated systemic symptoms from the review of systems. [1]
Smith and Nozik recommend a strategy called "name meshing." [2] The naming process involves extracting relevant clinical and historical information and creating a case profile (eg, acute unilateral nongranulomatous anterior uveitis in a 35-year-old white male with stiffness in the lower back). This information is meshed with clinical characteristics of known ocular inflammatory diseases. Using this information, the practitioner can generate a list of differential diagnoses and then order laboratory tests based on the likelihood that one of those diseases is present.
Although these systems work well, they are by no means foolproof. As the disease becomes chronic, the inflammation may change from nongranulomatous to granulomatous, the pattern of ocular involvement may change, and systemic features that were not initially present may become apparent. Because of the dynamic nature of uveitis, updating the review of systems and categorizing the ocular inflammation accordingly are important. Laboratory tests are no substitute for a systematic approach and careful observation in treating patients with uveitis.
Choosing the Correct Diagnostic Test
Ordering a standard series of tests in every patient with uveitis rarely is useful, can be costly, and often can confuse the diagnostic process. Rosenbaum and Wernick emphasize the futility of indiscriminate testing. [3] The value of the test varies with the probability of the suspected disease (pretest probability). The test is most helpful in raising diagnostic probability to rule in levels when the history and physical examination findings create a pretest probability of 40-60%. Bayes theorem can be used to assess the use of positive results by evaluating the sensitivity and specificity of a diagnostic test combined with the pretest probability of disease to calculate the posttest probability of disease.
Responsible clinical physicians decide which diagnostic tests to order for patients based on the best possible evidence. Sackett and colleagues coined the term evidence-based medicine, which is "the conscientious, explicit and judicious use of the current best evidence in making decisions about the care of individuals." [4] Evidence-based medicine is a lifelong learning process, incorporating sound clinical skills with a proficiency for a critical appraisal of the medical literature. The following section is based on the excellent work by Sackett and colleagues; the reader is encouraged to consult the References section. [4]
Evidence-based Medicine Approach to the Diagnosis of Uveitis
When a patient presents with intraocular inflammation, the diagnostic workup begins with the signs and symptoms. Each facet of the history and physical examination serves to either increase or decrease the probability of a target disorder being present. Unequivocally, the power of the clinical examination in establishing a diagnosis is far greater than the laboratory evaluation. Sandler discovered that after the history, a correct diagnosis was established in 56% of patients in a general medical clinic; it increased to 73% after the physical examination. [5] Routine laboratory evaluation helped to establish a diagnosis in only 5% of patients; not surprisingly, it also added substantial cost.
One strategy used by nearly all clinicians is the formulation of a list of potential diagnoses from a brief interaction with the patient, followed by a focused history and physical examination, and then an appropriate diagnostic evaluation that shortens the list of diagnostic possibilities. This strategy is termed the hypothetico-deductive approach by Sackett and colleagues. [4] In evaluating patients with uveitis, revisiting certain aspects of the history is common once particular facets of the examination become apparent. For example, after an initial interaction with a 34-year-old Black woman with symptoms of redness, pain, and photophobia, the clinician discovers that she has mutton-fat keratic precipitates and iris nodules. Further questioning is directed toward the various entities that cause granulomatous inflammation, with the appropriate diagnostic tests ordered accordingly.
To understand the value of a particular diagnostic test, the concepts of pretest and posttest probability, sensitivity, specificity, and likelihood ratios (LRs) need to be introduced. Pretest probability, defined as the odds that a given disease exists in a specific patient with a particular constellation of signs and symptoms, greatly influences which diagnostic tests are ordered. Very few published pretest probability values exist for the various uveitic entities. Consequently, they usually are derived from the practitioner's clinical experience and intuition. Alternatively, colleagues can be consulted about their experiences, thereby generating a pretest probability by consensus.
A diagnostic test is only useful if it can confidently rule in or rule out disease. In other words, a reliable test is one that is positive in nearly all patients with the disease, giving the clinician confidence that the individual is disease free when the test result is negative (for that disease).
Some commonly used parameters for determining whether the evidence for a diagnostic test is important include the sensitivity and specificity of the test and the newer, more powerful concept of LRs. Sensitivity is the proportion of patients with the target disorder who have a positive test result (true positives), whereas specificity represents the proportion of patients without the target disorder who have a negative test result (true negatives). By using Bayes theorem, the posttest probability of disease (ie, likelihood that a disease is present after a test result is known) can be calculated, given the sensitivity and specificity of the diagnostic test and the pretest probability of the disease.
Knowing the posttest probability provides the clinician with objective evidence about a diagnostic conclusion or determines whether the clinician pursues further laboratory evaluation. Calculating the posttest probability from Bayes theorem is cumbersome and not always practical. Fortunately, an easier method using a nomogram, which interprets a diagnostic test result by using a newer concept (ie, LRs), is available. This attribute of a particular test indicates the probability that a given test result would be expected in a patient with the target disorder, compared to the probability that the same result would be present in a patient without the target disorder. Any valid article on diagnostic testing should provide information regarding the sensitivity and specificity of the test, and, if that is all that is given, then the LR can be calculated easily.
The LR for a positive test result is LR+ = sensitivity/1-specificity; the LR for a negative test result is LR- = 1-sensitivity/specificity. For example, a patient presents with retinal vasculitis and other signs and symptoms suggestive of Wegener granulomatosis. Literature is searched, and an article about the use of antineutrophil cytoplasmic antibodies (C-ANCA) with a cytoplasmic staining pattern in testing for Wegener granulomatosis is found. According to the article, 90% of patients with biopsy-proven disease had a positive C-ANCA (sensitivity), and 10% of patients had a positive test but other causes of systemic vasculitis (false positives or 1-specificity). Therefore, in this example, the LR+ is .9/.1 or 9. In other words, the positive C-ANCA in this patient is 9 times more likely to be observed in an individual with Wegener granulomatosis than without the disease.
By consulting the Fagan nomogram (conveniently located in the pocket guide Evidence-Based Medicine: How to Practice and Teach EBM by Sackett et al), the posttest probability of disease can be calculated. [4] For example, if a patient has a 50% chance of being diagnosed with the disease before the test, the LR of 9 translates into a posttest probability of 92%, suggesting that the C-ANCA was a very useful test.
More and more investigators are realizing the value of the LR as a measure of diagnostic accuracy and incorporating it in their reports. In addition to calculating the posttest probability, the LR has other advantages over the sensitivity and specificity; it is less likely to vary with the prevalence of the target disease, and it can be calculated for several levels of the diagnostic test, symptom, or sign (ie, very positive to weakly positive).
Ultimately, the usefulness of a diagnostic test is determined by whether it helps doctors in caring for their patients (ie, test results changing the management of patients with uveitis, patients benefiting as a result of the test). In the above example, the diagnosis of Wegener granulomatosis was crucial because treatment with cyclophosphamide favorably impacts the mortality rate and ocular complications. This section briefly introduced some concepts that are important to the diagnostic approach, particularly in diagnosing uveitis. The authors believe that implementing the principles of evidence-based medicine in the daily practice of medicine makes better clinicians and, more importantly, better doctors for patients.
Diagnostic Approach to Specific Uveitic Entities
In an effort to better organize the classification and grading of various uveitic entities, the Standardization of Uveitis Nomenclature (SUN) Working Group published a consensus report in 2005. [6] This report provides clarification of the anatomical classification of uveitis. The reader is referred to this document as the prevailing methodology for the classification, descriptors, and terminology in the care of patients with uveitis.
Anterior uveitis
Patients with anterior uveitis present with a wide range of symptoms. These symptoms vary from a mild blurring of the vision with an otherwise normal-looking eye (ie, juvenile idiopathic arthritis [JIA]) to severe pain, photophobia, and loss of vision associated with intense injection and hypopyon. Factors other than ocular signs and symptoms can help in diagnosing anterior uveitis. The onset, duration, and severity of any symptom, as well as unilaterality or bilaterality of the disease, should be known. The patient's age, racial background, and ocular history should be taken into consideration. A detailed history and review of systems are of immeasurable value in the diagnostic approach to patients with uveitis.
An important element of any classification system for uveitis is defining what part of the eye is involved. The presence of white cells confined solely to the anterior chamber is called iritis. When the cellular activity involves the retrolental vitreous, the inflammatory process is believed to include the ciliary body and iris and is known as iridocyclitis. Corneal or scleral involvement plus anterior chamber inflammation is called keratouveitis or sclerouveitis, respectively.
Multiple etiologies are noted for anterior uveitis. Most types of anterior uveitis are sterile inflammatory reactions, as opposed to many of the posterior uveitic syndromes that are caused by infections. The percentage of idiopathic anterior uveitis ranges from approximately 38% to more than 70%; this is by far the most common cause of anterior uveitis. [7, 8] The next most common etiology is the sudden-onset human leukocyte antigen (HLA)-B27–positive or HLA-B27–associated disease.
McCannel reports that both community-based patients and university-based patients have similar incidence rates (about 17%). [9] After that, differences are observed in the probability of the various etiologies, depending on the clinical setting. For community-based patients, trauma is the third most common cause of anterior uveitis (5.7%); trauma was not observed in the university setting. Although herpes simplex virus is uncommon in community-based patients (1.9%), it was the third most likely diagnosis in the university setting (12.4%). Varicella-zoster infection occasionally was observed in both settings (5-6%). A large population-based study in Taiwan found that patients diagnosed with herpes zoster or herpes zoster ophthalmicus had a relative risk of anterior uveitis 1.67 and 13.06 times, respectively, greater than the comparison cohort during the first year after diagnosis. [10]
Pain, redness, and photophobia comprise the classic presentation of acute anterior uveitis. The pain usually is described as a dull ache in and around the eye, but anterior uveitis can cause little or no discomfort. Vision can be normal or slightly decreased. Often, the eye is extremely sensitive to light (photophobia). The patient may notice redness in one or both eyes or no change at all in the look of the eye.
The conjunctiva classically shows perilimbal injection (known as ciliary flush). The cornea may have keratic precipitates, which are clusters of WBCs collected on the endothelium. The type of keratic precipitate can provide a clue to the classification of anterior uveitis. Mutton-fat keratic precipitates are characteristic of granulomatous uveitis. Diffuse stellate keratic precipitates classically are seen in Fuchs uveitis syndrome. Interstitial keratitis commonly is seen in patients with syphilis and herpetic disease.
By definition, the anterior chamber has variable amounts of white cells floating in the aqueous. Often, protein also is visible in the anterior chamber as flare. If enough white cells deposit on the bottom of the chamber, a hypopyon results. This finding is suggestive of HLA-B27 disease, Behçet disease, or endophthalmitis.
The intraocular pressure (IOP) is often low in acute cases of anterior uveitis (with the exception of herpetic uveitis) but may be elevated in chronic cases.
The iris can provide additional information about the possible etiology or chronicity of the disease. Long-standing inflammation can cause posterior synechiae. Inflammatory nodules on the iris suggest granulomatous uveitis. Heterochromia is the classic finding in Fuchs uveitis syndrome. Atrophy of the iris may point to herpes zoster as the infection responsible for the inflammation.
The lens may show signs of cataractous change, which may suggest repeated bouts of iritis, or inflammatory precipitates may be present on the anterior lens capsule.
The anterior vitreous may have some cells that have "spilled over" from the anterior chamber. Some HLA-B27 diseases have varying amounts of vitritis and posterior pole involvement.
Papillitis or disc edema may be seen in Vogt-Koyanagi-Harada (VKH) disease, sarcoidosis, tuberculosis, Lyme disease, multiple sclerosis, toxoplasmosis, and toxocariasis.
In terms of testing, HLA-B27 is a genotype located on the short arm of chromosome 6 and is sometimes associated with specific rheumatologic diseases. HLA-B27 is present in 1.4-8% of the general population; however, it is present in as many as 50-60% of patients with acute iritis. The HLA-B27 test should be considered in patients with recurrent anterior nongranulomatous uveitis. These so-called seronegative spondyloarthropathies are associated strongly with both acute anterior uveitis and a positive HLA-B27 test. By definition, patients with these disorders do not have a positive rheumatoid factor. Some examples include ankylosing spondylitis, Reiter syndrome, inflammatory bowel disease, psoriatic arthritis, and postinfectious arthritis.
A thorough review of systems frequently directs the clinician toward the correct diagnosis. The possibility always exists of other systemic inflammatory disorders, some of which can be cured or at least managed. Syphilis, tuberculosis, Lyme disease, and herpes viruses are infectious diseases that can present as an anterior uveitis.
History is important in determining risk factors, but laboratory evidence of the disease is necessary so proper antibiotic therapy can be initiated quickly. Sarcoidosis is a systemic disease that classically manifests as an anterior granulomatous uveitis but can present as any type of uveitis. Judicious use of laboratory tests should help to better define the etiology of any anterior uveitis.
Table 1 is provided as a guide to the various clinical scenarios that may be confronted by the clinician. Beginning with knowledge of the type of inflammation, eliciting some associated factors should lead to a possible disease. Then, confirmatory laboratory tests can be used to establish a diagnosis. In general, a workup is required if the anterior uveitis is bilateral, severe, recurrent, or granulomatous or if the posterior segment is involved. Minimal laboratory testing should include CBC count, urinalysis, ACE, Venereal Disease Research Laboratory (VDRL) test, and fluorescent treponemal antibody absorption (FTA-ABS) test. Chest radiography should also be performed.
Table 1. Various Clinical Scenarios Encountered by Practitioner of Uveitis (Open Table in a new window)
Type of Inflammation |
Associated Factors |
Suspected Disease |
Laboratory Tests |
Acute/sudden onset, severe with or without fibrin membrane or hypopyon |
Arthritis, back pain, GI/genitourinary symptoms |
Seronegative spondyloarthropathies |
HLA-B27, sacroiliac films |
Aphthous ulcers |
Behçet disease |
HLA-B5, HLA-B51 |
|
Postsurgical, posttraumatic |
Infectious endophthalmitis |
Vitreous tap, vitrectomy |
|
Medication induced |
Rifabutin |
None |
|
None |
Idiopathic |
Possibly HLA-B27 |
|
Moderate severity of pain and redness |
Shortness of breath, African descent, subcutaneous nodules |
Sarcoidosis |
Serum ACE, lysozyme, chest radiograph or chest CT scan, gallium scan, biopsy |
Posttraumatic |
Traumatic iritis |
... |
|
Increased IOP, sectorial iris atrophy, corneal dendrite |
Herpetic iritis |
... |
|
Poor response to steroid, manifestations of 2° or 3° syphilis, HIV |
Syphilis |
Rapid plasma reagent (RPR) or VDRL, FTA-ABS |
|
Postcataract extraction, white plaque on posterior capsule |
Endophthalmitis, intraocular lens (IOL)- related iritis |
Vitrectomy and/or culture, consider anaerobic and fungal cultures |
|
Medication induced |
Etidronate (Didronel), metipranolol (OptiPranolol), latanoprost (Xalatan) |
|
|
History of HIV, alcohol abuse, exposure to infected individuals, residence in endemic regions |
Tuberculosis |
Purified protein derivative (PPD), QuantiFERON®-TB Gold, chest radiograph, referral to infectious disease specialist |
|
None |
Idiopathic |
... |
|
Chronic, minimal signs of redness or pain |
Child, especially with arthritis |
JIA-related iridocyclitis |
Antinuclear antibody (ANA), erythrocyte sedimentation rate (ESR) |
Heterochromia, diffuse stellate keratic precipitate, unilateral |
Fuchs uveitis syndrome |
None |
|
Postsurgical |
Low-grade endophthalmitis, IOL |
Vitrectomy, capsulectomy with culture |
|
None |
Idiopathic |
Lyme titers (possibly) |
Intermediate uveitis
Intermediate uveitis is an anatomic term suggested by the SUN Working Group. Intermediate uveitis is defined as intraocular inflammation that predominantly involves the peripheral retina, pars plana, and vitreous. Other terms used in the literature include chronic cyclitis, peripheral uveitis, and pars planitis. The term pars planitis is reserved to describe a subgroup of patients with idiopathic intermediate uveitis with snowbanking and/or snowball formation.
Intermediate uveitis accounts for approximately 8-15% of patients with uveitis in tertiary referral centers in the United States. Because characterization of this disease (and terminology associated with it) has been ambiguous, the conclusions of some older epidemiologic studies have been called into question. However, the report by Rodriguez et al used IUSG criteria and found that 162 of 1237 patients (13%) had intermediate uveitis, essentially confirming other studies. [7]
Patients typically present with painless blurred vision and floaters. Photophobia and redness are unusual.
Ocular findings include mild-to-moderate anterior segment inflammation, although anterior cellular activity may be more pronounced in children and in patients with multiple sclerosis. Presence of anterior vitreous cells is the sin qua non of this disorder, and, occasionally, the vitritis is severe enough to cause profound loss of vision. White clumps of inflammatory cells (called snowballs) tend to accumulate at the vitreous base where perivascular exudation and neovascularization may be present. The presence of a whitish yellow exudative material on the peripheral retina and the pars plana (called snowbanking) is commonly seen. The presence of this material facilitates the diagnosis but is not required to establish a diagnosis of intermediate uveitis. This finding is more consistent in patients with idiopathic intermediate uveitis and in children.
Because intermediate uveitis has been described in association with several systemic disorders, the initial diagnostic evaluation should exclude masquerade syndromes and infectious diseases in which immunosuppression may be ineffective or contraindicated. The diagnostic approach to intermediate uveitis should focus on the history and clinical examination. As stated by Henderly et al and also by Rodriguez et al, approximately two thirds of patients have idiopathic intermediate uveitis (pars planitis). [7, 11] Of the 162 patients with intermediate uveitis described by Rodriguez et al, 69% of them were idiopathic, sarcoidosis was diagnosed in 22% of them, multiple sclerosis was diagnosed in 8% of patients, and Lyme disease was diagnosed in only 1 patient. [7] Other entities have been reported to cause or to be confused with intermediate uveitis (see Table 2).
The authors' efforts focus on excluding sarcoidosis and multiple sclerosis with a thorough review of systems. Generally, the authors order an ACE level and a chest radiography for all patients to rule out subclinical sarcoidosis. The presence of neurologic symptoms or a history of optic neuritis may necessitate an MRI of the brain with subsequent neurologic consultation to rule out multiple sclerosis.
Patients from endemic areas for Lyme disease with a history of a rash typical of erythema migrans, chronic arthritis, or cranial nerve palsies undergo testing for antibodies to Borrelia burgdorferi. The authors seek consultation with a gastroenterologist for those patients with symptoms suggestive of inflammatory bowel disease or Whipple disease (if the diagnosis has not already been established). Older patients presenting with vitreous cells may be indicative of intraocular large cell lymphoma. Diagnostic vitrectomy, cytological evaluation of cerebrospinal fluid (CSF), and neuroimaging may be necessary.
Table 2. Other Entities Reported to Cause or Confused With Intermediate Uveitis (Open Table in a new window)
Clinical Entity |
Diagnostic Tests |
Idiopathic (pars planitis) |
None |
Sarcoidosis |
ACE, chest radiography, gallium scan, biopsy (possibly) |
Multiple sclerosis |
MRI and neurologic consultation if history of neurologic symptoms or optic neuritis, HLA-DR2 |
Lyme disease |
Lyme serology (immunoglobulin [Ig]G/IgM Western immunoblot testing) if from endemic area and/or presence of systemic signs |
Syphilis |
VDRL, FTA-ABS |
Inflammatory bowel disease |
GI consultation |
Whipple disease |
GI consultation |
Lymphoma |
Vitreous cytology with immunophenotyping, lumbar puncture for cytology, neuroimaging |
Retinal vasculitis
Conditions causing retinal vasculitis are a heterogeneous group of disorders that include some of the most devastating medical diseases encountered by the ophthalmologist. The term vasculitis implies primary retinal vascular inflammation (ie, immune complex deposition due to type III hypersensitivity), as seen in Behçet disease, but vasculitis is commonly a sign of intraocular inflammation from other causes with secondary vascular involvement (eg, toxoplasmosis).
Some noninflammatory retinal vascular diseases can be associated with perivascular exudation, such as diabetic retinopathy, radiation retinopathy, sickle cell retinopathy, venous occlusive disease, and Coat disease. The differential diagnosis of retinal vasculitis can be subdivided broadly into those diseases that have systemic involvement and those diseases that are confined to the eye, as outlined below.
-
Systemic diseases associated with retinal vasculitis
Behçet disease
Syphilis
Systemic lupus erythematosus (SLE)
Tuberculosis
Sarcoidosis
Wegener granulomatosis
Toxoplasmosis
Cytomegalovirus (CMV)
Polyarteritis nodosa (PAN)
Candidiasis
Multiple sclerosis
Herpes zoster/herpes simplex
Giant cell arteritis
Lyme disease
Crohn disease
Rickettsia
Whipple disease
Large cell lymphoma
Polymyositis/dermatomyositis
-
Ocular diseases associated with retinal vasculitis
Eales disease
Frosted branch angiitis
Acute retinal necrosis
Retinal arteritis and aneurysms
Birdshot choroiditis
Toxoplasmosis
Pars planitis
Common presenting ocular symptoms include painless blurred vision or severe visual loss, floaters, and scotomata.
Examination reveals perivascular exudation or cuffing that predominantly involves the arteries, the veins, or both; varying degrees of anterior chamber cell and flare; and vitritis. These findings may be accompanied by retinal hemorrhages, cotton-wool spots, exudates, cystoid macular edema (CME), neovascularization, vitreous hemorrhage, or disc edema.
The history and clinical examination are by far the most powerful diagnostic tools in evaluating this complex group of diseases. After a thorough review of systems and physical examination, the lack of findings indicating a systemic disease makes the pretest probability of disease very low and, thus, reduces the predicative value of any diagnostic tests.
Some investigators have shown that patients with retinal vasculitis presenting with symptoms and signs confined to the eye needlessly undergo exhaustive and expensive diagnostic evaluation. When George et al reviewed a series of patients with primary retinal vasculitis, they found that 96% of them had a negative review of systems; however, all patients underwent an exhaustive battery of tests. [12] A diagnosis was established in only one patient; false-positive results were obtained in 21% of patients. The shotgun approach has no place in evaluating a patient with retinal vasculitis. It invariably results in false-positive results, which, in turn, leads to more unnecessary testing, cost, and inconvenience to the patient.
Fluorescein angiography is an important aspect of the evaluation process. Findings may include staining of the vessel walls, beaded vessels, microaneurysms, telangiectatic vessels, capillary nonperfusion, neovascularization, and CME. Fluorescein angiography helps to classify the vasculitic process as either occlusive or nonocclusive. Perhaps more importantly, fluorescein angiography aids in making the determination as to whether a noninflammatory retinal vascular disease is present.
Numerous tests are ordered for all patients with intraocular inflammation, primarily to rule out masquerade syndromes and to evaluate the status of the patient's health. Such a limited workup includes CBC count, urinalysis, FTA-ABS, ACE, and chest radiography. Tests for syphilis and sarcoidosis help to rule in or rule out these 2 readily treatable diseases, which can present as any type of intraocular inflammation. From here, aspects of the history and clinical examination are used to formulate a differential diagnosis. If the pretest probability of disease is sufficiently high (but not too high), then further testing should be performed accordingly or treatment initiated if a clinical diagnosis is established.
A flowchart summarizing the diagnostic approach to retinal vasculitis is provided in the image below.
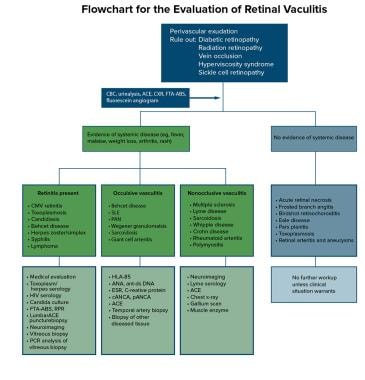 Flowchart for the evaluation of retinal vasculitis.
Flowchart for the evaluation of retinal vasculitis.
This chart is intended to serve as a guide for the thought process involved in approaching the diagnosis of retinal vasculitis. Implicit in this diagnostic approach is the need for consultation with an internist and possibly a rheumatologist or an infectious disease specialist. Most cases are idiopathic (sometimes called Eales disease), and many of the systemic diseases are clinical diagnoses (eg, Behçet disease, SLE). In these cases, laboratory tests are supportive but not diagnostic.
Inflammatory chorioretinopathies
This diverse group of choroidal inflammatory disorders is named because of their association with multiple, well-circumscribed, whitish yellow lesions affecting the choroid and choriocapillaris-retinal pigment epithelium (RPE) complex. Certain well-defined ocular inflammatory and infectious conditions, such as sarcoidosis, VKH disease, sympathetic ophthalmia, mycobacterium avium complex, and P carinii choroiditis, can be associated with well-circumscribed choroidal lesions. These clinical entities nearly always are associated with other ocular and extraocular manifestations, thereby posing little diagnostic dilemma. However, intraocular large cell lymphoma commonly presents with multiple punctate subretinal infiltrates that can be confused easily with an inflammatory process.
This discussion is limited to those disorders that are idiopathic and inflammatory in nature. The term white dot syndromes is intentionally vague because of the lack of understanding of their pathogenesis. Consequently, most of the diseases have descriptive terms attached to them, such as evanescent and placoid, implying ignorance as to their etiology. Some of these entities have overlapping clinical findings, prompting a debate as to whether they represent parts of a spectrum of a single disease or whether they are distinct clinical disorders.
These diseases are all clinical diagnoses; therefore, the clinical presentation and, to a larger degree, the ocular findings are vital in the diagnostic evaluation. Several of these diseases have an acute onset (eg, multiple evanescent white dot syndrome [MEWDS], acute posterior multifocal placoid pigment epitheliopathy [APMPPE]), whereas others are insidious in onset (eg, birdshot retinochoroiditis). Most of these diseases have little, if any, anterior or vitreous cellular activity, except birdshot retinochoroiditis, which can be associated with significant vitritis (without snowbanking), retinal vasculitis, and CME. A fundus finding, such as a granular appearance of the macula, is pathognomonic for MEWDS, whereas the lesions of other diseases occasionally can be confused with each other. The authors have found fluorescein angiography to be a helpful test in evaluating these patients.
Table 3 outlines clinical features and tests that are useful in the diagnostic approach to white dot syndromes.
Laboratory investigation of these patients is uniformly unproductive with the exception of birdshot retinochoroiditis. The association between this disease and the HLA-A29 phenotype is very strong. Testing for the HLA-A29 antigen is both sensitive and specific and has a relative risk of 132 to 157. Patients with a moderately high pretest probability of disease benefit from HLA typing because the result may rule in or rule out disease.
Table 3. Clinical Features and Tests Useful in Diagnostic Approach to White Dot Syndromes (Open Table in a new window)
|
Clinical Presentation |
Cellular Activity |
Fundus Lesions |
Fluorescein Angiography |
Other Tests |
Birdshot retinochoroiditis |
Insidious onset, middle age, F>M, bilateral (90%) |
Mild anterior chamber cells, mild-to-moderate vitritis |
100-300 µm, cream colored, indistinct, posterior pole to equator |
Early hyperfluorescence, macular and disc leakage |
HLA-A29+ (90%), electroretinography (ERG) findings abnormal |
APMPPE |
Acute onset, ± viral prodrome, young, M=F, bilateral, ± CNS symptoms |
1+ anterior chamber cells, ± mild vitreous cells |
Large, flat, cream colored, placoid, primarily in posterior pole |
Early hypofluorescence with late staining |
Cerebrospinal fluid (CSF) pleocytosis, ERG and electroretinography (EOG) findings negative |
MEWDS |
Acute onset, young, F>M, unilateral (90%), ± viral prodrome |
± mild vitreous cells |
Many, discrete, white, 100-200 µm, posterior pole, granular macula |
Wreath pattern of hyperfluorescence with late staining |
ERG and EOG findings very abnormal |
Serpiginous choroidopathy |
Insidious onset, middle age, M=F, bilateral |
± Mild anterior chamber and vitreous cells |
Large geographic, starts peripapillary with helicoid progression |
Window defects, loss of choriocapillaris, acute lesion shows blocked fluorescence |
... |
Punctate inner choroidopathy (PIC) |
Insidious onset, young, myopic, F>M, bilateral |
Quiet |
100-300 µm, white, punctate, posterior pole |
Block early, stain late |
ERG and EOG findings normal |
Multifocal choroiditis with panuveitis (MCP) |
Insidious onset, young, myopic, F>M, bilateral (80%) |
± Mild anterior chamber cells, vitreous cells (100%) |
100-300 µm, multifocal, punched-out |
Early hyperfluorescence, late staining |
ERG and EOG findings normal |
Acute retinal pigment epitheliitis (ARPE) |
Acute onset, young, M=F, unilateral (75%) |
Quiet |
Small black spots with halo around fovea |
Early blockage with halo of hyperfluorescence and late staining |
EOG findings negative |
Surgical Treatment of Uveitis
Background
Surgical indications in the management of uveitis include visual rehabilitation, diagnostic biopsy when findings may change the treatment plan, and removal of media opacities to monitor the posterior segment. Despite advances in anti-inflammatory and immunomodulatory therapy, permanent structural changes can occur in the eye that are best managed with surgery (eg, cataract formation, secondary glaucoma due to pupillary block or angle closure, retinal detachment).
In preparing the eye for surgery, medical treatment should be intensified for a minimum of 3 months to achieve complete quiescence of inflammation (ie, complete eradication of anterior chamber cells, active vitreous cells). Generally, beginning 24-48 hours preoperatively, topical prednisolone acetate 1% is administered every 1-2 hours (while awake) with prednisone (1 mg/kg) depending on the nature of the inflammation. Intraocular and/or periocular steroids may be administered intraoperatively. Dexamethasone intravitreal implant (Ozurdex) is approved and indicated to treat noninfectious uveitis affecting the posterior segment of the eye and may be considered to replace multiple daily dosing that is required for ophthalmic corticosteroids. Systemic and topical medications are tapered slowly postoperatively depending on the degree of inflammation.
Table 4. Specific Surgical Conditions Possibly Encountered by Practitioner of Uveitis (Open Table in a new window)
Condition |
Common Etiologies |
Surgical Procedure |
Comment |
Corneal opacification |
Herpetic keratitis, peripheral ulcerative keratitis |
Penetrating keratoplasty |
High risk of rejection or recurrence |
Band keratopathy |
Juvenile idiopathic arthritis (JIA) |
Chelation, excimer laser |
May require general anesthesia |
Cataract |
Any type of uveitis |
Phacoemulsification ± intraocular lens (IOL), vitrectomy and lensectomy |
See below (Cataract surgery in patient with uveitis) |
IOL precipitates |
Chronic anterior uveitis |
YAG laser "polishing" |
May be recurrent and require long-term topical steroid use |
Pupillary membranes |
JIA |
YAG laser, pars plana vitrectomy (PPV) |
High YAG laser energy may exacerbate uveitis |
Glaucoma, pupillary block, 2° angle closure |
Chronic anterior uveitis, herpetic uveitis, Fuchs heterochromic iridocyclitis |
Laser iridectomy, filtering surgery with mitomycin vs aqueous drainage tube |
Consider performing laser peripheral iridectomy when 270° or more of posterior synechiae |
Vitreous opacification |
Intermediate uveitis, sarcoidosis, vitreous hemorrhage, intraocular lymphoma |
PPV |
See indications for vitrectomy in patient with uveitis. |
Epiretinal membrane |
Intermediate uveitis, any posterior uveitis |
PPV/membrane peeling |
See indications for vitrectomy in patient with uveitis. |
Retinal detachment |
Intermediate uveitis, cytomegalovirus (CMV) retinitis, acute retinal necrosis (ARN) |
PPV ± scleral buckle, long-acting gas, or silicone oil |
See indications for vitrectomy in patient with uveitis. |
Chronic CME |
Any type of uveitis |
PPV (possibly), intravitreal triamcinolone and/or bevacizumab |
Quality of evidence is weak |
Choroidal neovascular membrane |
Multifocal choroiditis, punctate inner choroiditis, ocular histoplasmosis syndrome |
Thermal laser (extrafoveal), photodynamic therapy (ocular histoplasmosis syndrome [OHS]), intravitreal bevacizumab, submacular surgery |
Trial of aggressive anti-inflammatory or immunosuppressive therapy first if possible |
Retinal/optic disc neovascularization |
Sarcoidosis, intermediate uveitis, Behçet disease |
Photocoagulation, cryopexy, intravitreal bevacizumab |
May respond to corticosteroids |
Progressive unresponsive chorioretinal lesions of unknown etiology |
Neoplastic and infectious diseases |
Chorioretinal biopsy |
Referral to institutions familiar with procedure and expertise in interpretation of specimen |
Medically unresponsive intermediate uveitis |
Pars planitis, sarcoidosis |
Pars plana cryopexy or laser photocoagulation PPV |
Double freeze thaw to area of pars plana exudate |
Indications for vitrectomy in patient with uveitis
Posterior and intermediate uveitis may be associated with significant vitreous opacification that is unresponsive to medical therapy. Visually disabling opacities may occur with intermediate uveitis. Retinal or optic disc neovascularization may complicate conditions associated with vasculitis or vascular occlusion (eg, pars planitis, Behçet disease, sarcoidosis), resulting in vitreous hemorrhage. Modern vitrectomy offers a therapeutic option in these situations.
Mediators of intraocular inflammation stimulate fibrous tissue proliferation, thus predisposing the eye to epiretinal membrane formation. No prospective clinical trials in the literature exist comparing outcomes of membrane peeling in eyes with uveitis and those with idiopathic causes. However, Dev et al reported favorable visual results in a group of patients with pars planitis. [13] The investigators also noted a postoperative improvement in the level of vitritis in all patients. Because the macula may already have irreversible damage (eg, scaring, chronic cystoid macular edema [CME], macular hole, capillary nonperfusion), proper patient selection and realistic patient expectations are important.
Conditions resulting in large areas of thin and atrophic retina (eg, CMV retinitis, ARN) commonly are complicated by retinal detachment. These types of retinal detachments usually are associated with atrophic posterior breaks. Vitrectomy with long-acting gas or silicone tamponade commonly is used with or without scleral buckling. Scleral buckling alone usually is not successful.
At times, potentially life-threatening malignant processes or infectious uveitis may be mistaken for immune-mediated intraocular inflammation. A thorough review of systems and knowledge of response to prior therapy are critical. Vitrectomy or needle aspiration for diagnostic and therapeutic reasons is indicated whenever the intraocular inflammation responds poorly or incompletely to appropriate therapy or if clinical suspicion is raised for intraocular neoplasia or infection. Common masquerade syndromes in patients with vitritis include primary intraocular lymphoma, chronic fungal or anaerobic endophthalmitis, and retinal detachment. Whenever the diagnosis of lymphoma is entertained, the threshold to perform vitrectomy should be low; it is better to have a high number of negative biopsy results rather than to miss the diagnosis of this lethal condition. Proper specimen retrieval and handling and communication with the cytopathologist or microbiology laboratory are critical.
Several reports are available in the ophthalmic literature (all of them are retrospective and uncontrolled) that address the issue of therapeutic vitrectomy as a means of moderating intraocular inflammation. Vitrectomy may debulk the antigenic stimulus in the eye, thus reducing intraocular inflammation and CME, and allow tapering or elimination of systemic therapy. While the theoretical advantages of vitrectomy in chronic uveitis seem apparent, and although the body of evidence is growing, the quality of the evidence is weak. The role of vitrectomy in the management of uveitis will likely expand to include the placement of intraocular sustained-release drug delivery devices as well as to modulate the inflammatory response. Likewise, as polymerase chain reaction (PCR) becomes more widely used, diagnostic vitrectomy will be performed more regularly to confirm or establish a diagnosis and to guide therapy.
Cataract surgery in patient with uveitis
Cataracts occur in most patients with chronic or recurrent uveitis likely due to inflammatory mediators and corticosteroid-induced mechanisms. Indications for cataract surgery include the following: (1) visually significant lens opacity with good visual potential, (2) in combination with vitreoretinal surgery to better visualize the posterior segment, (3) lens-induced uveitis, and (4) impairment of fundus assessment. Prognosis is strongly dependent on preoperative control of all intraocular inflammation for a minimum of 3 months but preferably 3 to 6 months. Use aggressive topical, periocular, and oral steroid regimens in combination with immunomodulatory therapy if needed. Bringing the eye into a state of quiescence is key to a favorable outcome.
Modern techniques have improved surgical outcomes. Evidence suggests that phacoemulsification in conjunction with small incision surgery causes less breakdown of the blood-aqueous barrier compared with extracapsular techniques. Similarly, other investigators have found that clear corneal incisions are less likely to cause early postoperative inflammation than sclerocorneal tunnels. The optimal lens biomaterial has yet to be found, but some evidence exists in the literature that heparin-modified IOLs may provoke less inflammation than other materials. Until a superior biomaterial is discovered, placement of an all polymethyl methacrylate (PMMA) lens in the capsular bag with attention to meticulous cortical cleanup is recommended.
Cataract extraction and IOL placement is probably safe in patients with Fuchs heterochromic iridocyclitis, pars planitis, acute posterior multifocal placoid pigment epitheliopathy (APMPPE), ocular histoplasmosis, and chronic well-controlled anterior uveitis. Consider vitrectomy/lensectomy in patients with JIA, panuveitis, and granulomatous diseases. Placement of an IOL in children with JIA probably should be avoided, although some reports offer encouraging results with IOL implantation in this otherwise complicated form of uveitis.
Medical Treatment of Uveitis
Traditionally, medical management consisted of topical or systemic corticosteroids and often cycloplegics. In patients with severe cases of uveitis who were unresponsive to steroids or in those patients with complications associated with the usual therapy, immunosuppressants can be used. Immunosuppressive agents should be considered as first-line therapy in patients with Behçet disease involving the posterior segment, Wegener granulomatosis, and necrotizing scleritis. These diseases often are associated with life-threatening systemic vasculitis, and available evidence suggests that treatment with immunosuppressive agents can improve outcomes in these diseases. [14]
TNF inhibitors
Immunomodulatory therapy often is used in situations where long-term treatment with systemic corticosteroids is necessary, such as serpiginous choroiditis, birdshot choroiditis, Vogt-Koyanagi-Harada (VKH) disease, sympathetic ophthalmia, and juvenile idiopathic arthritis (JIA).
The newest entries into the therapeutic arena for uveitis are medications that target specific mediators of the immune response. Although these medications have been studied primarily in patients with rheumatoid arthritis, psoriasis, and Crohn disease, similarities in the disease pathogenesis have stimulated interest in using these drugs for the treatment of various ocular inflammatory diseases. In particular, molecules that block the tumor necrosis factor alpha (eg, adalimumab, infliximab) have been found to effectively modulate the immune response in patients with uveitis. [15, 16]
In June 2016, the FDA approved adalimumab (Humira) for the treatment of noninfectious intermediate uveitis, posterior uveitis, and panuveitis in adults. Approval was based on results from two pivotal phase 3 studies, VISUAL-I and VISUAL-II, which demonstrated that adult patients with active and controlled noninfectious intermediate uveitis, posterior uveitis, and panuveitis had a significantly lower risk for treatment failure, a combination of uveitic flare and decrease in visual acuity, when treated with adalimumab compared with placebo. [17, 18]
The VISUAL-I study found that compared to placebo, patients on adalimumab were less likely to experience treatment failure (P< 0.001). Median time to treatment failure was prolonged by 87%, from 3 months for placebo to 5.6 months for adalimumab. [17] In the VISUAL-II study, the median time to treatment failure was 8.3 months for placebo and not estimable for adalimumab, as more than half of the adalimumab-treated patients did not experience treatment failure (P=0.004). [18]
Infliximab has been used investigationally in the treatment of refractory uveitis, but, in one study, the favorable effects were mitigated by a relatively high incidence of drug toxicity and possibly a loss of efficacy with long-term use. [19] A more recent study has shown adalimumab and infliximab to have comparable efficacy and incidence of serious adverse effects. [20]
Intraocular implants and intravitreal injections
Another new treatment modality is the use of intraocular pharmacotherapy via intravitreal injection and surgically placed implants. Several reports have confirmed the treatment benefit of intravitreal triamcinolone (4 mg/0.1 mL or 4 mg/0.05 mL) for the management of refractory cystoid macular edema (CME). Unfortunately, the intravitreal half-life of triamcinolone is relatively short, and multiple injections may be necessary. Cataract formation and elevated intraocular pressure (IOP) are common, and the risk of endophthalmitis (usually sterile) is approximately 0.1%.
Reports are emerging regarding the off-label use of the full-length humanized anti-vascular endothelial growth factor (VEGF) monoclonal antibody bevacizumab in the treatment of refractory CME and neovascular complications of uveitis. [21] However, as with triamcinolone, serial injections may be necessary, and the long-term tolerability and safety of this medication is unknown.
Sustained-release fluocinolone intravitreal implants (Retisert, Yutiq) are approved by the US Food and Drug Administration (FDA) for the treatment of refractory noninfectious uveitis. Retisert is released at an initial rate of about 0.6 mcg/day during the first month, and then at 0.3-0.4 mcg/day for 30 months. It was shown to effectively controlled inflammation in nearly all eyes in the phase 3 study, allowing for the tapering of systemic steroids and immunomodulatory agents. [22] A second fluocinolone ophthalmic implant (Yutiq) was approved in 2018; it releases about 0.25 mcg/day over 36 months. Cataract formation is a near certainty in phakic eyes, and the risk of glaucoma is nearly 60%. Careful patient selection is a must.
The results of the 2-year Multicenter Uveitis Steroid Treatment (MUST) Trial, a randomized controlled trial comparing a 3-year fluocinolone implant with conventional systemic immunosuppressive therapy, showed no statistically significant difference in vision at 24 months between the 2 treatment arms. However, the implant group achieved faster and more complete control of inflammation and less vitreous haze, but this was at the expense of cataract formation in nearly all phakic eyes and elevated intraocular pressure in two thirds of eyes. [23]
One randomized controlled trial found that insertion of an intravitreal dexamethasone implant modulated inflammation and improved vision at 26 weeks in patients with intermediate or posterior uveitis. [24]
Cycloplegics
Symptoms and complications of inflammation can be tempered with topical cycloplegic agents. Both short-acting drops (eg, cyclopentolate) and long-acting drops (eg, atropine) can be used to decrease photophobia caused by ciliary spasm and to break up or prevent the formation of posterior synechiae.
Cyclopentolate (I-Pentolate, Cyclogyl, AK-Pentolate) prevents muscle of the ciliary body and sphincter muscle of the iris from responding to cholinergic stimulation. It induces mydriasis in 30 to 60 minutes and cycloplegia in 25 to 75 minutes. Effects last up to 24 hours.
Atropine ophthalmic (Isopto) acts at parasympathetic sites in smooth muscle to block response of the sphincter muscle of iris and muscle of ciliary body to acetylcholine, causing mydriasis and cycloplegia.
Corticosteroids
Corticosteroids inhibit arachidonic acid release from phospholipids, inhibit the transcription and action of cytokines, and limit B- and T-cell activity. They are indicated in inflammatory diseases of a noninfectious cause. Several routes of ocular administration are available (eg, topical, intravitreal, periocular, systemic, and suprachoroidal). The best route and dose is determined for each patient, but the minimum amount needed to control inflammation should be used to reduce complications. Because of serious adverse effects, especially with high doses and long-term use, immunosuppressive agents commonly are used for chronic or sight-threatening uveitis.
Topical: For anterior uveitis, topical steroid drops are used. Depending on the severity of the inflammation being treated, the frequency can range from hourly to every other day. Prednisolone acetate 1% is the most commonly used agent, but difluprednate 0.05% is more potent and may be more efficacious. Prednisolone acetate is a precipitate; therefore, it is important that the patient vigorously shake the bottle before use. However, since difluprednate is an emulsion, shaking is unnecessary. Sometimes, steroids can cause ocular hypertension; therefore, patients must be monitored at 4- to 6-week intervals.
Intravitreal implants: Dexamethasone and fluocinolone intravitreal inserts may be considered as an option to provide sustained treatment.
Periocular: When a more posterior effect is necessary or when compliance is an issue, periocular corticosteroids can be administered. Either a transseptal or a sub-Tenon approach works to deposit a long-lasting steroid (eg, triamcinolone acetonide) around the eye. Initially treating patients with a topical steroid for 3 to 4 weeks prior to the administration of a long-acting depot steroid may help to identify those patients who are steroid responders. Some evidence exists that deep transseptal injections cause less ocular hypertension than the sub-Tenon method. These injections should not be used in patients with infectious uveitis or scleritis because scleral thinning and possible perforation could result.
Systemic: When systemic disease is present that also requires treatment or for vision-threatening uveitis that is poorly responsive to other methods of delivery, oral or intravenous therapy is necessary. Both the short-term and long-term adverse effects of corticosteroid use should be discussed with the patient and may require the help of an internist. Prednisone is the most commonly used oral corticosteroid.
Prednisolone ophthalmic (Pred Forte) treats acute inflammation following eye surgery or other types of insults to eye. It decreases inflammation and corneal neovascularization. It also suppresses migration of polymorphonuclear leukocytes and reverses increased capillary permeability. In cases of bacterial infections, concomitant use of anti-infective agents is mandatory; if signs and symptoms do not improve after 2 days, reevaluate the patient. Dosing may be reduced, but advise patients not to discontinue therapy prematurely.
Dexamethasone intravitreal implant (Ozurdex) delivers the corticosteroid to the eye in a sustained implant. This is indicated for chronic noninfectious uveitis.
Fluocinolone ophthalmic (Retisert) delivers the corticosteroid to the eye in a sustained implant. This is also indicated for chronic noninfectious uveitis.
Triamcinolone intravitreal (Trivaris, Triesence) decreases inflammation by suppressing migration of polymorphonuclear leukocytes and reversing capillary permeability. Trivaris and Triesence are triamcinolone products that are preservative-free and specifically indicated for intravitreal administration. A 20- to 40-mg sub-Tenon or transseptal injection is recommended and may be repeated in 2-3 weeks.
Triamcinolone suprachoroidal (Xipere) was approved by the FDA in October 2021 for macular edema associated with uveitis. This administration provides more precisely located corticosteroid therapy to the affected posterior areas of the eye. This decreases the area of the eye exposed to corticosteroids, thus decreasing the risk of adverse effects locally and systemically. A drug dose reduction is also an advantage due the high bioavailability of the drug in the choroid.
Approval was supported by results of the phase 3 PEACHTREE clinical trial (n = 160) showing an improvement in best-correct visual activity of 15 letters or more at week 24 and continued improvement throughout a 24 week follow-up period compared to placebo (p < 0.01). Trial also showed higher proportion of patients experienced clinically meaningful reductions in central subfield thickness compared to placebo. [25]
Prednisone (Deltasone, Orasone, Meticorten, Sterapred) may be used if topical therapy is not adequate to treat iritis (especially in bilateral cases). It may decrease inflammation by reversing increased capillary permeability and suppressing PMN activity.
Immunosuppressive agents
Immunosuppressive agents include three main categories of therapy: antimetabolites, T-cell suppressors, and cytotoxic agents. Antimetabolites include azathioprine, methotrexate, and mycophenolate mofetil. T-cell inhibitors include cyclosporine and tacrolimus. Cytotoxic agents are alkylating agents and include cyclophosphamide and chlorambucil. Most agents take several weeks to achieve efficacy; therefore, they initially are used in conjunction with oral corticosteroids. Once the disease is under control, corticosteroids can be tapered. Instituting these agents and monitoring of adverse events in conjunction with a specialist who has expertise with these agents is strongly recommended.
Azathioprine (Imuran) is a nucleoside analog that interferes with DNA replication and RNA transcription. It decreases peripheral T-lymphocyte and B-lymphocyte count and reduces lymphocyte activity. Metabolism depends on xanthine oxidase. It may decrease proliferation of immune cells, which results in lower autoimmune activity. Azathioprine is indicated to treat Behçet disease or chronic uveitis, especially with oral corticosteroids.
Methotrexate (Rheumatrex, Folex PFS) is a folic acid analog and inhibitor of dihydrofolate reductase, which is the enzyme responsible for the conversion of dihydrofolate to tetrahydrofolate. It arrests DNA replication, inhibiting rapidly dividing cells (eg, leukocytes). It is eliminated primarily through the kidney. Methotrexate is used to treat various ocular inflammatory diseases, including vasculitis, panuveitis, intermediate uveitis, and vitritis.
Mycophenolate (CellCept) is a selective inhibitor of inosine monophosphate dehydrogenase, which interferes with guanosine nucleotide synthesis. It prevents lymphocyte proliferation, suppresses antibody synthesis, interferes with cellular adhesion to vascular endothelium, and decreases recruitment of leukocytes to sites of inflammation. It is metabolized primarily through the kidneys. Mycophenolate is used in combination with other agents, particularly oral corticosteroids and may be an acceptable alternative to azathioprine or methotrexate, especially in patients intolerant of other agents.
Cyclosporine (Neoral, Sandimmune) is an inhibitor of transcription in T lymphocytes that are in the G0 and G1 phase of their cell cycle, which blocks replication and ability to produce lymphokines. It is metabolized in liver. Cyclosporine is useful as sole therapy for various uveitis conditions.
Tacrolimus (Prograf) is a naturally produced macrolide immunosuppressant that suppresses humoral immunity (T lymphocyte) activity. It is metabolized by the cytochrome P-450 system. Small, uncontrolled case series suggested that it might be effective for treating noninfectious uveitis.
Cyclophosphamide (Neosar, Cytoxan) is chemically related to nitrogen mustards. As an alkylating agent, the mechanism of action of the active metabolites may involve cross-linking of DNA, which may interfere with growth of normal and neoplastic cells. It is cytotoxic to resting and dividing lymphocytes. It is primarily excreted through kidney.
Chlorambucil (Leukeran) is an alkylating agent that substitutes an alkyl group for hydrogen ions in organic compounds. DNA-to-DNA intrastrand cross-linking and DNA-to-protein cross-linking occur, which lead to interference in DNA replication and transcription. Metabolism occurs in the liver. Small studies suggest it may be effective for various sight-threatening uveitic syndromes, including Behçet disease and sympathetic ophthalmia.
-
Flowchart for the evaluation of retinal vasculitis.
Tables
- Table 1. Various Clinical Scenarios Encountered by Practitioner of Uveitis
- Table 2. Other Entities Reported to Cause or Confused With Intermediate Uveitis
- Table 3. Clinical Features and Tests Useful in Diagnostic Approach to White Dot Syndromes
- Table 4. Specific Surgical Conditions Possibly Encountered by Practitioner of Uveitis
Type of Inflammation |
Associated Factors |
Suspected Disease |
Laboratory Tests |
Acute/sudden onset, severe with or without fibrin membrane or hypopyon |
Arthritis, back pain, GI/genitourinary symptoms |
Seronegative spondyloarthropathies |
HLA-B27, sacroiliac films |
Aphthous ulcers |
Behçet disease |
HLA-B5, HLA-B51 |
|
Postsurgical, posttraumatic |
Infectious endophthalmitis |
Vitreous tap, vitrectomy |
|
Medication induced |
Rifabutin |
None |
|
None |
Idiopathic |
Possibly HLA-B27 |
|
Moderate severity of pain and redness |
Shortness of breath, African descent, subcutaneous nodules |
Sarcoidosis |
Serum ACE, lysozyme, chest radiograph or chest CT scan, gallium scan, biopsy |
Posttraumatic |
Traumatic iritis |
... |
|
Increased IOP, sectorial iris atrophy, corneal dendrite |
Herpetic iritis |
... |
|
Poor response to steroid, manifestations of 2° or 3° syphilis, HIV |
Syphilis |
Rapid plasma reagent (RPR) or VDRL, FTA-ABS |
|
Postcataract extraction, white plaque on posterior capsule |
Endophthalmitis, intraocular lens (IOL)- related iritis |
Vitrectomy and/or culture, consider anaerobic and fungal cultures |
|
Medication induced |
Etidronate (Didronel), metipranolol (OptiPranolol), latanoprost (Xalatan) |
|
|
History of HIV, alcohol abuse, exposure to infected individuals, residence in endemic regions |
Tuberculosis |
Purified protein derivative (PPD), QuantiFERON®-TB Gold, chest radiograph, referral to infectious disease specialist |
|
None |
Idiopathic |
... |
|
Chronic, minimal signs of redness or pain |
Child, especially with arthritis |
JIA-related iridocyclitis |
Antinuclear antibody (ANA), erythrocyte sedimentation rate (ESR) |
Heterochromia, diffuse stellate keratic precipitate, unilateral |
Fuchs uveitis syndrome |
None |
|
Postsurgical |
Low-grade endophthalmitis, IOL |
Vitrectomy, capsulectomy with culture |
|
None |
Idiopathic |
Lyme titers (possibly) |
Clinical Entity |
Diagnostic Tests |
Idiopathic (pars planitis) |
None |
Sarcoidosis |
ACE, chest radiography, gallium scan, biopsy (possibly) |
Multiple sclerosis |
MRI and neurologic consultation if history of neurologic symptoms or optic neuritis, HLA-DR2 |
Lyme disease |
Lyme serology (immunoglobulin [Ig]G/IgM Western immunoblot testing) if from endemic area and/or presence of systemic signs |
Syphilis |
VDRL, FTA-ABS |
Inflammatory bowel disease |
GI consultation |
Whipple disease |
GI consultation |
Lymphoma |
Vitreous cytology with immunophenotyping, lumbar puncture for cytology, neuroimaging |
|
Clinical Presentation |
Cellular Activity |
Fundus Lesions |
Fluorescein Angiography |
Other Tests |
Birdshot retinochoroiditis |
Insidious onset, middle age, F>M, bilateral (90%) |
Mild anterior chamber cells, mild-to-moderate vitritis |
100-300 µm, cream colored, indistinct, posterior pole to equator |
Early hyperfluorescence, macular and disc leakage |
HLA-A29+ (90%), electroretinography (ERG) findings abnormal |
APMPPE |
Acute onset, ± viral prodrome, young, M=F, bilateral, ± CNS symptoms |
1+ anterior chamber cells, ± mild vitreous cells |
Large, flat, cream colored, placoid, primarily in posterior pole |
Early hypofluorescence with late staining |
Cerebrospinal fluid (CSF) pleocytosis, ERG and electroretinography (EOG) findings negative |
MEWDS |
Acute onset, young, F>M, unilateral (90%), ± viral prodrome |
± mild vitreous cells |
Many, discrete, white, 100-200 µm, posterior pole, granular macula |
Wreath pattern of hyperfluorescence with late staining |
ERG and EOG findings very abnormal |
Serpiginous choroidopathy |
Insidious onset, middle age, M=F, bilateral |
± Mild anterior chamber and vitreous cells |
Large geographic, starts peripapillary with helicoid progression |
Window defects, loss of choriocapillaris, acute lesion shows blocked fluorescence |
... |
Punctate inner choroidopathy (PIC) |
Insidious onset, young, myopic, F>M, bilateral |
Quiet |
100-300 µm, white, punctate, posterior pole |
Block early, stain late |
ERG and EOG findings normal |
Multifocal choroiditis with panuveitis (MCP) |
Insidious onset, young, myopic, F>M, bilateral (80%) |
± Mild anterior chamber cells, vitreous cells (100%) |
100-300 µm, multifocal, punched-out |
Early hyperfluorescence, late staining |
ERG and EOG findings normal |
Acute retinal pigment epitheliitis (ARPE) |
Acute onset, young, M=F, unilateral (75%) |
Quiet |
Small black spots with halo around fovea |
Early blockage with halo of hyperfluorescence and late staining |
EOG findings negative |
Condition |
Common Etiologies |
Surgical Procedure |
Comment |
Corneal opacification |
Herpetic keratitis, peripheral ulcerative keratitis |
Penetrating keratoplasty |
High risk of rejection or recurrence |
Band keratopathy |
Juvenile idiopathic arthritis (JIA) |
Chelation, excimer laser |
May require general anesthesia |
Cataract |
Any type of uveitis |
Phacoemulsification ± intraocular lens (IOL), vitrectomy and lensectomy |
See below (Cataract surgery in patient with uveitis) |
IOL precipitates |
Chronic anterior uveitis |
YAG laser "polishing" |
May be recurrent and require long-term topical steroid use |
Pupillary membranes |
JIA |
YAG laser, pars plana vitrectomy (PPV) |
High YAG laser energy may exacerbate uveitis |
Glaucoma, pupillary block, 2° angle closure |
Chronic anterior uveitis, herpetic uveitis, Fuchs heterochromic iridocyclitis |
Laser iridectomy, filtering surgery with mitomycin vs aqueous drainage tube |
Consider performing laser peripheral iridectomy when 270° or more of posterior synechiae |
Vitreous opacification |
Intermediate uveitis, sarcoidosis, vitreous hemorrhage, intraocular lymphoma |
PPV |
See indications for vitrectomy in patient with uveitis. |
Epiretinal membrane |
Intermediate uveitis, any posterior uveitis |
PPV/membrane peeling |
See indications for vitrectomy in patient with uveitis. |
Retinal detachment |
Intermediate uveitis, cytomegalovirus (CMV) retinitis, acute retinal necrosis (ARN) |
PPV ± scleral buckle, long-acting gas, or silicone oil |
See indications for vitrectomy in patient with uveitis. |
Chronic CME |
Any type of uveitis |
PPV (possibly), intravitreal triamcinolone and/or bevacizumab |
Quality of evidence is weak |
Choroidal neovascular membrane |
Multifocal choroiditis, punctate inner choroiditis, ocular histoplasmosis syndrome |
Thermal laser (extrafoveal), photodynamic therapy (ocular histoplasmosis syndrome [OHS]), intravitreal bevacizumab, submacular surgery |
Trial of aggressive anti-inflammatory or immunosuppressive therapy first if possible |
Retinal/optic disc neovascularization |
Sarcoidosis, intermediate uveitis, Behçet disease |
Photocoagulation, cryopexy, intravitreal bevacizumab |
May respond to corticosteroids |
Progressive unresponsive chorioretinal lesions of unknown etiology |
Neoplastic and infectious diseases |
Chorioretinal biopsy |
Referral to institutions familiar with procedure and expertise in interpretation of specimen |
Medically unresponsive intermediate uveitis |
Pars planitis, sarcoidosis |
Pars plana cryopexy or laser photocoagulation PPV |
Double freeze thaw to area of pars plana exudate |