Background
Lowe syndrome, also called oculocerebrorenal syndrome (OCRS) and oculocerebrorenal syndrome of Lowe (OCRL), is an X-linked recessive metabolic disorder described by Lowe and coworkers in 1952. [1] It is a multisystem disorder that primarily affects the eyes, nervous system, and kidneys. It is characterized by congenital cataracts, infantile glaucoma, neonatal or infantile hypotonia, intellectual impairment, and renal tubular dysfunction (Fanconi syndrome).
Oculocerebrorenal syndrome is so named because of the prominent involvement of the 3 major organ systems; however, involvement of bone, gonads, muscle, skin, and connective tissue, as well as stereotypic behavior, also can occur. Additional manifestations include corneal keloids, growth retardation, areflexia, metabolic acidosis, proteinuria, aminoaciduria, and noninflammatory arthropathy. These patients often exhibit the characteristic facial appearance of frontal bossing, deep set eyes, and full cheeks, although the characteristic phenotype is often difficult to identify in neonates.
Female carriers manifest characteristic lens opacities, but they typically have normal renal and neurologic function. See the image below.
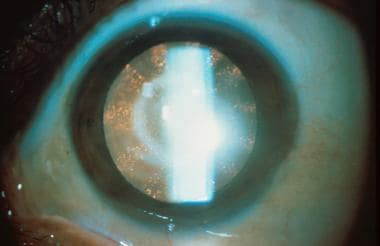 Classic lenticular opacities in a female carrier for Lowe syndrome. Note the punctate cortical opacities in radical wedges. Image courtesy of Otis Paul, MD.
Classic lenticular opacities in a female carrier for Lowe syndrome. Note the punctate cortical opacities in radical wedges. Image courtesy of Otis Paul, MD.
Pathophysiology
Lowe syndrome is caused by a mutation of the OCRL1 gene mapped to the chromosomal locus of Xq26.1. [2] The gene encodes a phosphatidylinositol (4,5) bisphosphate 5 phosphatase, localized to the trans-Golgi complex involved in actin polymerization. [2, 3] It is a key enzyme in the cellular phosphoinositide pathway, important in cellular trafficking. Deficiency of the enzyme causes the protean manifestations of Lowe syndrome. The reduced enzyme activity results in increased enzyme substrate and abnormal actin-binding proteins important in neuronal morphogenesis. [4, 5] Fibroblast activity was found to be decreased by 80%-90% in patients with Lowe syndrome compared to healthy controls. [6]
Epidemiology
Frequency
International
The estimated prevalence of Lowe syndrome is 1 per 500,000 population. The Lowe Syndrome Association (LSA) reported 190 males living with Lowe syndrome in 2000. [7] As of 2005, the Italian Association of Lowe’s Syndrome reported 34 individuals with Lowe syndrome living in Italy. [6] No cases of Lowe syndrome have been reported in Africa, South America, and parts of Asia. [6, 7]
Mortality/Morbidity
Ocular: The hallmark feature is congenital cataracts. Left untreated, the cataracts will cause nystagmus and eventual blindness. Glaucoma develops in 50-70% of patients, typically manifesting in the first year of life, but it may present at any age. [8] Patients also may develop corneal leukomas. Amblyopia may occur secondary to poor compliance with treatment and secondary strabismus. Corrected visual acuity is infrequently better than 20/70 despite optimal management. The visual impairment represents a combination of the morphologic changes in the eye, retinal dysfunction, and cortical functioning.
Renal: If untreated, the proximal renal tubular acidosis leads to failure to thrive and metabolic collapse. By the second to third decade, gradual loss of creatinine clearance occurs, with progressive renal failure.
Growth: At birth, these patients are within the normal growth curve but fall off in length, height, and weight in subsequent years.
Neurological: Neonatal hypotonia may contribute to poor feeding and delayed motor development.
Life expectancy: Patients with appropriate therapy may live to be 30-40 years of age, generally dying from renal failure, respiratory distress, status epilepticus, or infection. With improved medical treatment, infection as a cause for death has declined dramatically. The expected life span has not yet been defined.
Sex
Lowe syndrome occurs almost exclusively in males. It is an X-linked recessive disorder. The female carrier state is identifiable by characteristic lenticular opacities.
A few cases have been reported in females, often having X-autosome translocations involving the OCRL1 locus, explained by the inactivation of the normal X chromosome by the translocation. [2]
Age
Lowe syndrome is congenital.
Prognosis
Currently, there is no correlation between the genotype and phenotypes, so clinical severity of disease cannot be correlated with certain variants. However, affected males within the same family have similar phenotypes, as intrafamilial variability has not yet been reported. [6]
There is complete penetrance. [7]
The oldest reported patient to live with Lowe syndrome died at age 54 years. [2]
-
Classic lenticular opacities in a female carrier for Lowe syndrome. Note the punctate cortical opacities in radical wedges. Image courtesy of Otis Paul, MD.






