Background
First described by Gass in 1968, acute posterior multifocal placoid pigment epitheliopathy (APMPPE) is an acquired inflammatory disorder affecting the retina, retinal pigment epithelium, and choroid of otherwise young healthy adults. [1] The disease is self-limited and is characterized by multiple yellow-white placoid subretinal lesions of the posterior pole, as shown below. The lesions are frequently bilateral and in various stages of evolution, typically resolving in weeks to months and leaving circumscribed areas of retinal pigment epithelial disturbance.
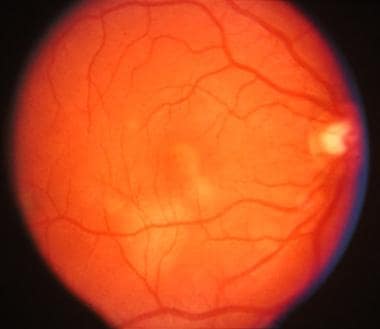 Posterior pole of right eye. Early acute posterior multifocal placoid pigment epitheliopathy lesion shows yellowish-white placoid lesion involving the macula and an area just inferior temporal to the macula.
Posterior pole of right eye. Early acute posterior multifocal placoid pigment epitheliopathy lesion shows yellowish-white placoid lesion involving the macula and an area just inferior temporal to the macula.
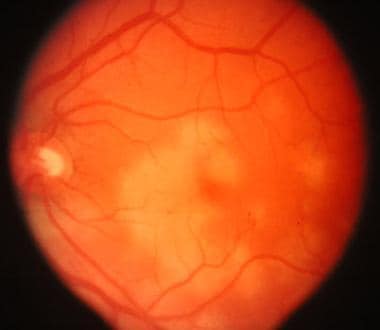 Posterior pole of left eye of same patient showing acute posterior pole placoid lesion.
Posterior pole of left eye of same patient showing acute posterior pole placoid lesion.
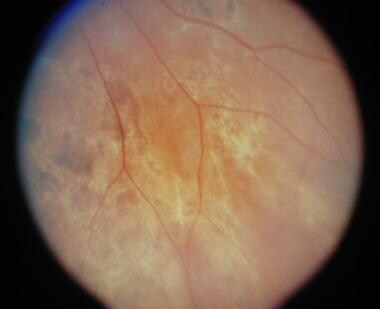 Inferior nasal of right eye of the same patient approximately 2 months later, showing scattered areas of retinal pigment epithelium atrophy and hyperplasia.
Inferior nasal of right eye of the same patient approximately 2 months later, showing scattered areas of retinal pigment epithelium atrophy and hyperplasia.
Synonyms of APMPPE include acute multifocal placoid pigment epitheliopathy (AMPPE), acute placoid pigment epitheliopathy, and multifocal placoid pigment epitheliopathy.
Pathophysiology
The pathophysiology of APMPPE is still inconclusive owing to a lack of histopathological evidence of acute disease activity.
Gass initially suggested that inflammation of the retinal pigment epithelium (RPE) and outer retina manifests as placoid lesions during the acute phase based on clinical and fluorescein angiography (FA) findings. [1]
Growing evidence supports choriocapillary perfusion abnormalities in both acute and healed phases as the underlying pathology in APMPPE, with secondary changes in the RPE and outer retina. Nonetheless, an understanding of the exact pathogenesis is still elusive owing to interpretative difficulties with new imaging technologies and lack of histopathology. [2]
Epidemiology
Frequency
United States
The incidence and prevalence of APMPPE is unknown. However, since the landmark description of Gass, APMPPE has been reported frequently and widely from ophthalmic centers, primarily in the United States and Western Europe. Most US patients reported in the literature reside in the northern and midwestern states.
International
Reports in the international literature have included patients from northwestern European countries, such as England, Scotland, France, Belgium, the Netherlands, and Denmark. Several reports have originated from Japan. Reports from other geographic areas have been sparse.
Mortality/Morbidity
APMPPE usually affects healthy adults, and, other than ocular involvement, systemic manifestations are relatively uncommon. When they do occur, many systemic manifestations are usually mild and transient in nature; however, the presence of cerebral vasculitis has been associated with permanent neurologic sequelae, such as hemiparesis and even death from intracerebral edema and brain herniation in rare instances.
Race
Almost 80% of the recorded cases include whites, with the remainder being Japanese, African American, Nepalese, and from the Indian subcontinent. Whether this racial distribution represents a predilection of APMPPE for whites or a reporting bias is unclear.
Sex
Earlier reports of APMPPE suggested a slight preponderance of women with this disease, but more recent publications suggest no sexual predilection, with equal frequency between both men and women.
Age
The mean age of onset is approximately 27 years. The documented age range of onset is 7-66 years. The most frequent age range of occurrence of APMPPE is in those patients aged 16-40 years, representing approximately 85% of cases. About 50% of patients present in the third decade of life.
Prognosis
APMPPE is a self-limiting disease, with more than 80% of affected individuals achieving a visual acuity of 20/40 or better. Visual recovery may take months. In some cases, patients with foveal involvement end up with a visual acuity of 20/50 or worse. [3]
A small number of patients have permanent visual loss due to choroidal neovascularization.
A few patients have long-term functional ocular symptoms (eg, scotomata, metamorphopsia).
An occasional death has been reported following an episode of cerebral vasculitis.
Other ocular and systemic manifestations of vasculitis usually are self-limited and non–life threatening.
Chorioretinal scarring usually is associated with few visual symptoms. [4]
Patient Education
Reassure the patient that in spite of significant vision loss, the visual decrease is usually transient and many patients regain relatively good vision.
-
Posterior pole of right eye. Early acute posterior multifocal placoid pigment epitheliopathy lesion shows yellowish-white placoid lesion involving the macula and an area just inferior temporal to the macula.
-
Fluorescein angiography showing peripheral hypofluorescence and central leakage of the lesion inferior temporal to the macula.
-
Posterior pole of left eye of same patient showing acute posterior pole placoid lesion.
-
Fluorescein angiogram of the patient above in late phase showing late staining of placoid areas.
-
Inferior nasal of right eye of the same patient approximately 2 months later, showing scattered areas of retinal pigment epithelium atrophy and hyperplasia.
-
Fluorescein angiography of same patient in late phase showing areas of late staining.
-
This image shows localized scleritis superiorly.





