Practice Essentials
Stevens-Johnson syndrome is a type IV (subtype C) hypersensitivity reaction that typically involves the skin and the mucous membranes. Although several classification schemes have been reported, the simplest classification breaks the disease down as follows [1] :
-
Stevens-Johnson syndrome: A minor form of toxic epidermal necrolysis, with less than 10% body surface area (BSA) detachment
-
Overlapping Stevens-Johnson syndrome/toxic epidermal necrolysis: Detachment of 10-30% of the BSA
-
Toxic epidermal necrolysis: Detachment of more than 30% of the BSA
Signs and symptoms
Typical prodromal symptoms of Stevens-Johnson syndrome are as follows:
-
Cough productive of a thick, purulent sputum
-
Headache
-
Malaise
-
Arthralgia
Patients may complain of a burning rash that begins symmetrically on the face and the upper part of the torso. The cutaneous lesions are characterized as follows:
-
The rash can begin as macules that develop into papules, vesicles, bullae, urticarial plaques, or confluent erythema
-
The typical lesion has the appearance of a target; this is considered pathognomonic
-
In contrast to the typical lesions of erythema multiforme, these lesions have only 2 zones of color
-
The lesion’s core may be vesicular, purpuric, or necrotic; that zone is surrounded by macular erythema
-
Lesions may become bullous and later rupture, leaving denuded skin; the skin becomes susceptible to secondary infection
-
Urticarial lesions typically are not pruritic
-
Infection may be responsible for the scarring associated with morbidity
-
Although lesions may occur anywhere, the palms, soles, dorsum of the hands, and extensor surfaces are most commonly affected
-
The rash may be confined to any one area of the body, most often the trunk
Signs of mucosal involvement can include the following:
-
Erythema
-
Edema
-
Sloughing
-
Blistering
-
Ulceration
-
Necrosis
The following ocular signs may be noted on slit-lamp examination:
-
Eyelids: Trichiasis, distichiasis, meibomian gland dysfunction, blepharitis
-
Conjunctiva: Papillae, follicles, keratinization, subepithelial fibrosis, conjunctival shrinkage, foreshortening of fornices, symblepharon, ankyloblepharon
-
Cornea: Superficial punctate keratitis, epithelial defect, stromal ulcer, neovascularization, keratinization, limbitis, conjunctivalization, stromal opacity, perforation (see the image below)
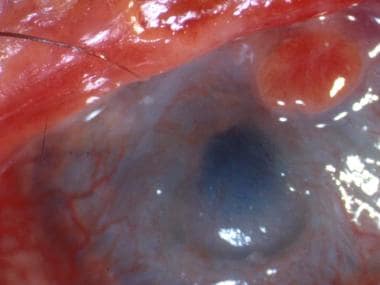 A patient with severe eye involvement associated with Stevens-Johnson syndrome. Note corneal neovascularization and conjunctivalization of the ocular surface.
A patient with severe eye involvement associated with Stevens-Johnson syndrome. Note corneal neovascularization and conjunctivalization of the ocular surface.

See Clinical Presentation for more detail.
Diagnosis
Minimal dermal inflammatory cell infiltrate and full-thickness necrosis of the epidermis are typical histopathologic findings in patients with Stevens-Johnson syndrome. Histopathologic examination of the skin can also reveal the following:
-
Changes in the epidermal-dermal junction ranging from vacuolar alteration to subepidermal blisters
-
Dermal infiltrate: Superficial and mostly perivascular
-
Apoptosis of keratinocytes
-
CD4+ T lymphocytes predominating in the dermis; CD8+ T lymphocytes predominating in the epidermis; the dermoepidermal junction and epidermis is infiltrated mostly by CD8+ T lymphocytes
Ocular examination can demonstrate the following:
-
Conjunctival biopsies from patients with active ocular disease show subepithelial plasma cells and lymphocyte infiltration; lymphocytes also are present around vessel walls; the predominant infiltrating lymphocyte is the helper T cell
-
Immunohistology of the conjunctiva reveals numerous HLA-DR–positive cells in the substantia propria, vessel walls, and epithelium
See Workup for more detail.
Management
Most patients with Stevens-Johnson syndrome are treated symptomatically. In principle, the symptomatic treatment of patients with this disorder does not differ from the therapy applied to patients with extensive thermal or chemical burns.
Patients should be treated with special attention to airway and hemodynamic stability, fluid status, wound/burn care, and pain control. Therapy for Stevens-Johnson syndrome proceeds as follows:
-
Withdrawal of any agent suspected of causing the condition is critically important
-
Oral lesions are managed with mouthwashes; topical anesthetics are useful in reducing pain and allowing the patient to take in fluids
-
Areas of denuded skin must be covered with compresses of saline or Burow solution (an aqueous solution of aluminium triacetate)
-
Tetanus prophylaxis must be addressed
Extensive debridement of nonviable epidermis followed by immediate cover with biologic dressings is among the recommended treatments.
Ocular therapy
The treatment of acute ocular manifestations usually begins with aggressive lubrication of the ocular surface. As inflammation and cicatricial changes ensue, most ophthalmologists use topical steroids, antibiotics, and mechanical symblepharon lysis.
In the case of mild chronic superficial keratopathy, long-term lubrication may be sufficient. In cases of severe ocular involvement, treatment includes the following:
-
Removal of keratinized plaques from the posterior lid margins
-
Mucous membrane grafting and/or amniotic membrane grafting
-
Limbal stem cell transplantation and amniotic membrane grafting
-
Superficial keratectomy removing conjunctivalized or keratinized ocular surface
See Treatment and Medication for more detail.
Background
Stevens-Johnson syndrome (SJS) is a type IV (subtype C) hypersensitivity reaction that typically involves the skin and the mucous membranes. While minor presentations may occur, significant involvement of oral, nasal, eye, vaginal, urethral, gastrointestinal, and lower respiratory tract mucous membranes may develop during the illness. GI and respiratory involvement may progress to necrosis. Stevens-Johnson syndrome is a serious systemic disorder with the potential for severe morbidity and even death.
The syndrome was first described in 1922, when the American pediatricians Albert Mason Stevens and Frank Chambliss Johnson reported the cases of 2 boys aged 7 and 8 years with "an extraordinary, generalized eruption with continued fever, inflamed buccal mucosa, and severe purulent conjunctivitis." Both cases had been misdiagnosed by primary care physicians as hemorrhagic measles.
Erythema multiforme (EM), originally described by Ferdinand von Hebra in 1866, was part of the differential diagnosis in both cases but was excluded because of the "character of skin lesions, the lack of subjective symptoms, the prolonged high fever, and the terminal heavy crusting." Despite the presence of leukopenia in both cases, Stevens and Johnson in their initial report suspected an infectious disease of unknown etiology as the cause.
In 1950, Thomas divided EM into 2 categories: erythema multiforme minor (more common and as previously described by von Hebra) and erythema multiforme major (EMM). Since 1983, erythema multiforme major and Stevens-Johnson syndrome had been considered synonymous.
In the 1990s, however, Bastuji and Roujeau each proposed that erythema multiforme major and Stevens-Johnson syndrome are 2 distinct disorders. [2] They suggested that the denomination of erythema multiforme should be restricted to patients with typical targets or raised edematous papules, with or without mucosal involvement. This clinical picture is in accordance with the original description by von Hebra.
Bastuji and Roujeau further proposed that the denomination of Stevens-Johnson syndrome should be used for a syndrome characterized by mucous membrane erosions and widespread small blisters that arise on erythematous or purpuric maculae that are different from classic targets.
According to this clinical classification, erythema multiforme major and Stevens-Johnson syndrome could be 2 distinct disorders with similar mucosal erosions, but different patterns of cutaneous lesions. This hypothesis is supported further by a strong correlation between clinical classification and the probable cause.
Conversely, several investigators propose that Stevens-Johnson syndrome and toxic epidermal necrolysis (TEN) represent the same disease at different levels of severity. A unifying classification of "acute disseminated epidermal necrosis" or "exanthematic necrolysis" has been suggested.
Although several classification schemes have been reported, the simplest breaks the disease down as follows [1] :
-
Stevens-Johnson syndrome - A "minor form of TEN," with less than 10% body surface area (BSA) detachment
-
Overlapping Stevens-Johnson syndrome/toxic epidermal necrolysis (SJS/TEN) - Detachment of 10-30% BSA
-
Toxic epidermal necrolysis - Detachment of more than 30% BSA
An argument against this unifying concept was that HSV infection had been described as a frequent cause of Stevens-Johnson syndrome/erythema multiforme major but not of toxic epidermal necrolysis. However, reports showed that HSV infection has not definitively been related to Stevens-Johnson syndrome and suggested that clinical manifestations and pathology results support the linking of Stevens-Johnson syndrome and toxic epidermal necrolysis together with their differentiation from erythema multiforme.
Various etiologic factors (eg, infection, drugs, malignancies) have been implicated as causes of Stevens-Johnson syndrome. However, as many as half of cases are idiopathic. There is strong evidence for a genetic predisposition to Stevens-Johnson syndrome provoked by certain drugs. (See Etiology.)
There are no specific laboratory studies (other than biopsy) that can definitively establish the diagnosis of Stevens-Johnson syndrome (see Clinical and Workup). No specific treatment of Stevens-Johnson syndrome is noted; most patients are treated symptomatically. In principle, the symptomatic treatment of patients with Stevens-Johnson syndrome does not differ from the treatment of patients with extensive burns. Withdrawal of the suspected offending agent is critically important. Immunomodulatory treatment is controversial. (See Treatment.)
For patient education information, see the Skin, Hair, and Nails Center, as well as Life-Threatening Skin Rashes.
Pathophysiology
An idiosyncratic, delayed hypersensitivity reaction has been implicated in the pathophysiology of Stevens-Johnson syndrome. Certain population groups appear more susceptible to develop Stevens-Johnson syndrome than the general population. Slow acetylators, patients who are immunocompromised (especially those infected with HIV [3, 4] ), and patients with brain tumors undergoing radiotherapy with concomitant antiepileptics are among those at most risk.
Slow acetylators are people whose liver cannot completely detoxify reactive drug metabolites. For example, patients with sulfonamide-induced toxic epidermal necrolysis have been shown to have a slow acetylator genotype that results in increased production of sulfonamide hydroxylamine via the P-450 pathway. These drug metabolites may have direct toxic effects or may act as haptens that interact with host tissues, rendering them antigenic. [5, 6]
Antigen presentation and production of tumor necrosis factor (TNF)–alpha by the local tissue dendrocytes results in the recruitment and augmentation of T-lymphocyte proliferation and enhances the cytotoxicity of the other immune effector cells. [7] A "killer effector molecule" has been identified that may play a role in the activation of cytotoxic lymphocytes. [8] The activated CD8+ lymphocytes, in turn, can induce epidermal cell apoptosis via several mechanisms, which include the release of granzyme B and perforin.
In 1997, Inachi et al demonstrated perforin-mediated apoptosis in patients with Stevens-Johnson syndrome. [9] Perforin, a pore-making monomeric granule released from natural killer cells and cytotoxic T lymphocytes, kills target cells by forming polymers and tubular structures not unlike the membrane attack complex of the complement system.
Apoptosis of keratinocytes also can take place as a result of ligation of their surface death receptors with the appropriate molecules. Those can trigger the activation of the caspase system, leading to DNA disorganization and cell death. [10]
Apoptosis of keratinocytes can be mediated via direct interaction between the cell-death receptor Fas and its ligand. Both can be present on the surfaces of the keratinocytes. Alternatively, activated T-cells can release soluble Fas ligand and interferon-gamma, which induces Fas expression by keratinocytes. [1] Researchers have found increased levels of soluble Fas ligand in the sera of patients with SJS/TEN before skin detachment or onset of mucosal lesions. [11]
The death of keratinocytes causes separation of the epidermis from the dermis. Once apoptosis ensues, the dying cells provoke recruitment of more chemokines. This can perpetuate the inflammatory process, which leads to extensive epidermal necrolysis. [12]
Higher doses and rapid introduction of allopurinol [13] and lamotrigine [14] may also increase the risk of developing SJS/TEN. Risk is lessened by starting these at the low doses and titrating gradually. [15]
There is evidence that systemic lupus is a risk factor as well. [16]
Etiology
Various etiologic factors have been implicated as causes of Stevens-Johnson syndrome. Drugs most commonly are blamed. The 4 etiologic categories are as follows:
-
Infectious
-
Drug-induced
-
Malignancy-related
-
Idiopathic
Stevens-Johnson syndrome is idiopathic in 25-50% of cases. Drugs and malignancies are most often implicated as the etiology in adults and elderly persons. Pediatric cases are related more often to infections.
Infectious causes
Viral diseases that have been reported to cause Stevens-Johnson syndrome include the following:
-
Herpes simplex virus (possibly; remains a debated issue)
-
AIDS
-
Coxsackie viral infections
-
Influenza
-
Hepatitis
-
Mumps
In children, Epstein-Barr virus and enteroviruses have been identified as being associated with the development of Stevens-Johnson syndrome. More than half of the patients with Stevens-Johnson syndrome report a recent upper respiratory tract infection.
Bacterial etiologies include the following:
-
Group A beta-hemolytic streptococci
-
Diphtheria
-
Brucellosis
-
Lymphogranuloma venereum
-
Mycobacteria
-
Rickettsial infections
-
Tularemia
-
Typhoid
Possible fungal causes include coccidioidomycosis, dermatophytosis, and histoplasmosis. Malaria and trichomoniasis have been reported as protozoal causes.
Drug-induced
Antibiotics are the most common cause of Stevens-Johnson syndrome, followed by analgesics, cough and cold medication, NSAIDs, psycho-epileptics, and antigout drugs. Of antibiotics, penicillins and sulfa drugs are prominent culprits; ciprofloxacin also has been reported. [19]
The following anticonvulsants have been implicated:
-
Phenytoin
-
Carbamazepine
-
oxcarbazepine (Trileptal)
-
Valproic acid
-
Lamotrigine
-
Barbiturates
Mockenhaupt et al stressed that most anticonvulsant-induced SJS occurs in the first 60 days of use. [20]
Antiretroviral drugs implicated in Stevens-Johnson syndrome include nevirapine and possibly other non-nucleoside reverse transcriptase inhibitors. [21] Indinavir has been mentioned.
Stevens-Johnson syndrome has also been reported in patients taking the following drugs:
-
Modafinil (Provigil)
-
Allopurinol [22]
-
Mirtazapine [23]
-
TNF-alpha antagonists (eg, infliximab, etanercept, adalimumab) [24]
-
Cocaine
-
Sertraline
-
Pantoprazole
-
Tramadol
Genetic factors
There is strong evidence for a genetic predisposition to severe cutaneous adverse drug reactions such as Stevens-Johnson syndrome. Carriage of the following human leukocyte antigens has been associated with increased risk:
-
HLA-B*1502
-
HLA-B*5801
-
HLA-B*44
-
HLA-A29
-
HLA-B12
-
HLA-DR7
-
HLA-A2
-
HLA-B*5801
-
HLA-A*0206
-
HLA-DQB1*0601
Certain of these HLA alleles are associated with an increased probability of developing Stevens-Johnson syndrome upon exposure to specific drugs. The US Food and Drug Administration (FDA) and Health Canada advise screening for HLA-B*1502 in patients of southeastern Asian ethnicity before starting treatment with carbamazepine. (The risk is much lower in other ethnic populations, making screening impractical in them). HLA-B*5801 confers a risk of allopurinol-related reactions. [25] Pretreatment screening is not readily available. [26]
Whites with HLA-B*44 appear to be more susceptible to develop Stevens-Johnson syndrome. HLA-A29, HLA-B12, and HLA-DR7 are frequently associated with sulfonamide-induced Stevens-Johnson syndrome, while HLA-A2 and HLA-B12 are often encountered in Stevens-Johnson syndrome induced by nonsteroidal anti-inflammatory drugs (NSAIDs).
HLA-A*0206 and HLA-DQB1*0601 allele have been shown to be was strongly associated with Stevens-Johnson syndrome with ocular disease. [27, 28]
Nevertheless, whether the presence of those genes constitutes a predisposition to Stevens-Johnson syndrome or whether those genes are in linkage disequilibrium with more relevant adjacent genes is unknown. [29]
Epidemiology
Strom et al reviewed Medicaid billing data from 1980-1984 in Michigan, Minnesota, and Florida to determine the incidence of Stevens-Johnson syndrome; the incidence rates were 7.1, 2.6, and 6.8 cases per million population per year, respectively. [30]
Cases tend to have a propensity for the early spring and winter.
For overlapping SJS and TEN, oxicam NSAIDs (piroxicam, meloxicam, tenoxicam) and sulfonamides are most commonly implicated in the United States and other western nations. [26]
SJS occurs with a worldwide distribution similar in etiology and occurrence to that in the United States. However, a study from Germany reported only 1.1 cases per 1 million person-years.
In contrast to the drugs most often implicated in western nations, allopurinol is the most common offending agent in Southeast Asian nations, including Malaysia, Singapore, Taiwan, and Hong Kong. [26]
Race-, sex-, and age-related demographics
Stevens-Johnson syndrome has been described worldwide in all races, although it may be more common in Whites. Interestingly, disease is not limited to humans; cases have been reported in dogs, cats, and monkeys.
The proportion of females has been estimated to be 33-62%. The largest series reports 39.9% of females in a group of 315 patients with Stevens-Johnson syndrome.
In a large cohort, the mean age of patients with Stevens-Johnson syndrome was 25 years. In a smaller series, the mean age of patients with Stevens-Johnson syndrome was reported as 47 years. However, cases have been reported in children as young as 3 months and adults as old as 78 years.
Prognosis
Individual lesions typically should heal within 1-2 weeks, unless secondary infection occurs. Most patients recover without sequelae.
Mortality is determined primarily by the extent of skin sloughing. When body surface area (BSA) sloughing is less than 10%, the mortality rate is approximately 1-5%. However, when more than 30% BSA sloughing is present, the mortality rate is between 25% and 35%, and may be as high as 50%. [31, 26] Bacteremia and sepsis appear to play a major role in increased mortality. [32]
The SCORTEN score (a severity-of-illness score for toxic epidermal necrolysis) calculates the risk for death in both SJS and TEN based on the following variables:
-
Age >40 years
-
Malignancy
-
Heart rate >120
-
Initial percentage of epidermal detachment >10%
-
Blood urea nitrogen (BUN) level >10 mmol/L
-
Serum glucose level >14 mmol/L
-
Bicarbonate level < 20 mmol/L
Each variable is assigned a value of 1 point. Mortality rates are as follows:
-
0-1 points, ≥3.2%
-
2 points, ≥12.1%
-
3 points, ≥35.3%
-
4 points, ≥58.3%
-
5 or more points, ≥90%
Other negative prognostic factors include persistent neutropenia (defined as neutropenia lasting more than 5 days), hypoalbuminemia (usually < 2 g/dL), and persistent azotemia.
In a survival analysis of a cohort of patients with either Stevens-Johnson syndrome or toxic epidermal necrolysis, Sekula et al found that the severity of the cutaneous reaction causing either of these disorders was a risk factor for mortality, but only during the first 90 days following reaction onset. [33] The investigators also found that serious comorbidities and age were risk factors for mortality after 90 days, but not beyond 1 year, past reaction onset. Mortality among patients was 23% at 6 weeks and 34% at 1 year.
Although some patients rapidly progress to lose very large areas of the epidermis in a matter of days, the process suddenly ceases in others and re-epithelialization begins a few days later. Predicting the course of disease in a given patient at the initial presentation is not possible. Re-epithelialization is usually complete within 3 weeks, but pressure and mucosal areas may remain eroded and crusted for 2 weeks or longer.
Survivors of Stevens-Johnson syndrome may experience numerous long-term sequelae; the most disabling are those of the eye. Cicatrization of conjunctival erosions may lead to the following:
-
Inverted eyelashes
-
Photophobia
-
A burning sensation in the eyes
-
Watery eyes
-
A keratitis siccalike syndrome
-
Corneal and conjunctival neovascularization
As many as 40% of survivors of toxic epidermal necrolysis have residual potentially disabling lesions that may cause blindness.
-
A patient with severe eye involvement associated with Stevens-Johnson syndrome. Note corneal neovascularization and conjunctivalization of the ocular surface.
-
Epithelial defect of the cornea with neovascularization and surface conjunctivalization.
-
Note extensive sloughing of epidermis from Stevens-Johnson syndrome. Courtesy of David F. Butler, MD.
-
Sheetlike desquamation on the foot in a patient with toxic epidermal necrolysis. Courtesy of Robert Schwartz, MD.
-
Hemorrhagic crusting of the mucous membranes in toxic epidermal necrolysis. Similar lesions are seen in Stevens-Johnson syndrome. Courtesy of Robert Schwartz, MD.
-
Note early cutaneous slough with areas of violaceous erythema.
-
Extensive sloughing on the face.
-
Note the presence of both 2-zoned atypical targetoid lesions and bullae.
-
Extensive blistering and sloughing on the back.
-
Extensive sloughing on the back.
-
Note extensive sloughing.
-
Low-power view showing full-thickness epidermal necrosis.








