Practice Essentials
Optic neuritis (ON) is a demyelinating inflammation of the optic nerve that often occurs in association with multiple sclerosis (MS) and, much less commonly, neuromyelitis optica (NMO). A gradual recovery of all or part of the visual acuity with time is characteristic of ON, [1] although permanent residual deficits in color vision and contrast and brightness sensitivity are common. [2]
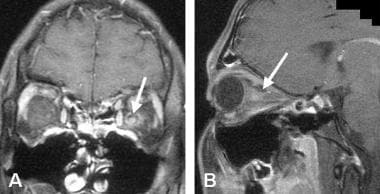 A case of acute optic neuritis. A. 1.5 Tesla, contrast-enhanced spin echo T1-weighted, fat-suppressed coronal MRI through the orbits shows enlargement and contrast enhancement of the left optic nerve in the retrobulbar portion (arrow). B. Coronal spin echo T1-weighted, fat-suppressed MRI of the same patient shows enlargement and contrast enhancement of the nerve in a parasagittal oblique section (arrow).
A case of acute optic neuritis. A. 1.5 Tesla, contrast-enhanced spin echo T1-weighted, fat-suppressed coronal MRI through the orbits shows enlargement and contrast enhancement of the left optic nerve in the retrobulbar portion (arrow). B. Coronal spin echo T1-weighted, fat-suppressed MRI of the same patient shows enlargement and contrast enhancement of the nerve in a parasagittal oblique section (arrow).
History
Classically, patients with ON are young, often are female, and have subacute vision loss associated with pain on eye movement.
The patient’s history may reveal the following signs and symptoms of optic neuritis:
-
Preceding viral illness
-
Rapidly developing impairment of vision in 1 eye or, much less commonly, both eyes [3]
-
Dyschromatopsia (change in color perception) in the affected eye, which may occasionally be more prominent than decreased vision [4]
-
Retro-orbital or ocular pain that occurs in association with the vision changes, which is usually exacerbated by eye movement and may precede vision loss
-
Uhthoff phenomenon, in which visual symptoms are exacerbated by heat or exercise
-
Pulfrich effect, in which objects moving in a straight line appear to have a curved trajectory when viewed with both eyes, presumably caused by asymmetric conduction between the optic nerves
Patients with MS may have recurrent attacks of ON, [5] which means that a history of episodes of decreased vision in the same or the fellow eye may be elicited.
Neuromyelitis optica is characterized by often severe, bilateral ON and myelitis in a close temporal relationship [6, 7, 8, 9] ; however, ON occasionally can precede the myelopathy.
Physical examination
Signs of ON may include the following:
-
Decreased pupillary light reaction in the affected eye: A relative afferent pupillary defect (RAPD) or Marcus Gunn pupil commonly is observed. When both eyes are affected, an RAPD may not be apparent.
-
Varying degrees of vision reduction: Vision reduction ranges from mildly decreased visual acuity to complete vision loss; it is usually monocular but may be binocular.
-
Abnormal contrast sensitivity and color vision are present in almost all adults with ON even in the absence of a measurable decrease in visual acuity.
-
Field defects: These may include altitudinal, arcuate, nasal step, central scotoma, and centrocecal scotoma.
-
Initially, the optic nerve head may appear normal, with disc pallor months later. Papillitis (swollen disc) may be seen in one third of patients with ON.
Diagnosis
The following blood tests can be performed when optic neuropathies other than demyelinating ON are suspected:
-
Erythrocyte sedimentation rate
-
Thyroid function tests
-
Antinuclear antibody test
-
Measurement of angiotensin-converting enzyme level
-
Rapid plasma reagin test
-
Mitochondrial DNA mutation studies
Magnetic resonance imaging (MRI) is highly sensitive for and specific in the assessment of inflammatory changes in the optic nerves; it is also specific for white matter lesions in the central nervous system. Magnetic resonance imaging also helps to rule out structural lesions. [10, 11]
Visual evoked potentials (VEPs) can be considered in patients in whom a diagnosis of ON is suspected. Visual evoked potentials may be abnormal even when visual acuity is normal and when MRI of the optic nerve reveals no abnormalities. Visual evoked potentials often show a loss of P100 response in the acute phase. With time, P100 recovers but usually continues to show a markedly prolonged latency that persists indefinitely, even after clinical recovery.
Management
For most patients with ON, treatment and recovery proceed as follows:
-
Visual function begins to improve 1 to several weeks after onset, even without any treatment.
-
Permanent residual deficits in color vision and contrast and brightness sensitivity are common. [2]
-
Pharmacologic therapy for patients with ON is directed toward ameliorating the acute symptoms of pain and decreased vision caused by demyelinating inflammation of the nerve; varying regimens of corticosteroids have been used for this purpose.
Eculizumab, a monoclonal antibody that targets C5, is the first drug specifically approved by the US Food and Drug Administration (FDA) for adults with neuromyelitis optica spectrum disorder (NMOSD) who are seropositive for anti-aquaporin-4 (AQP4) antibody. [15] The FDA has also approved inebilizumab, a monoclonal antibody that binds to CD19, [16] and satralizumab [Enspryng] for the treatment of NMOSD in adults who are seropositive for anti-aquaporin-4 or AQP4 antibody.
For patients with ON whose brain lesions on MRI indicate a high risk of developing clinically definite MS, treatment with immunomodulators (eg, interferonbeta-1a, interferon beta-1b, glatiramer acetate) may be considered. [17]
Background
Optic neuritis (ON) is a demyelinating inflammation of the optic nerve that typically first occurs in young adulthood. Many cases of ON are associated with multiple sclerosis (MS) or neuromyelitis optica (NMO), but ON can occur in isolation. [18] In cases associated with MS, ON is commonly the first manifestation of the chronic demyelinating process. [19] Long-term follow-up studies have indicated that up to 75% of female patients initially presenting with ON ultimately develop MS.
Occasionally, ON can result from an infectious process involving the orbits or paranasal sinuses or occur in the course of a systemic viral infection. [20, 21, 22, 23, 24, 25, 26, 27, 28] Certain optic neuropathies, such as anterior ischemic optic neuropathy (AION) or compressive and hereditary optic neuropathies, can resemble ON. [29]
This article reviews ON as a primary demyelinating inflammation of the nerve occurring either in isolation or in association with MS or NMO. (Neuromyelitis optica is a severe form of a demyelinating disease thought to be of autoimmune origin; it affects the optic nerves and the spinal cord, causing recurrent attacks of blindness and paralysis.) [30, 31] Much information has been gleaned from the Optic Neuritis Treatment Trial (ONTT), and the reader is encouraged to review the follow-up data from this study. [32, 33]
Etiology
Most cases of optic neuritis (ON) are associated with multiple sclerosis (MS), even though ON can occur in isolation. In MS-associated and isolated monosymptomatic ON, the cause is presumed to be an autoimmune reaction that results in a demyelinating inflammation of the nerve. Pathologic studies in patients with ON associated with MS have shown that the demyelinated lesions in the optic nerve are similar to the MS plaques seen in the brain, with an inflammatory response marked by perivascular cuffing, T cells, and plasma cells. However, little is known about the pathology of isolated ON.
In a single case of chronic, isolated ON, a biopsy specimen showed the presence of perivascular lymphocytic infiltration, multifocal demyelination, and reactive astrocytosis in the retrobulbar portion of the optic nerve. Abnormal intrathecal IgG synthesis, reflected as the presence of oligoclonal bands in the cerebrospinal fluid (CSF), is found in 60-70% of patients with isolated ON, suggesting an immunologic etiology similar to that of MS. [34]
Neuromyelitis optica (NMO) has been recognized as a distinct inflammatory demyelinating disease consisting of ON in combination with longitudinally extensive transverse myelitis. Neuromyelitis optica is associated with the presence of a specific serum, NMO IgG autoantibody, which targets the water channel aquaporin-4. [35, 36, 37]
Approximately two thirds or more of patients with neuromyelitis optica spectrum disorder (NMOSD) have IgG antibodies to aquaporin-4 (AQP4-IgG), a water channel protein that is abundant on astrocytic membranes and proximate to the blood-brain barrier. Patients who are seronegative cannot be distinguished clinically from those who are seropositive. In AQP4-IgG–seropositive disease, binding of AQP4-IgG to AQP4 on astrocytic end-feet initiates the activation of the complement cascade, the infiltration of granulocytes into the central nervous system, and antibody-dependent cell-mediated cytotoxicity.
Levels of interleukin-6 (IL-6) are elevated in the CSF of patients with NMOSD, as compared with patients who have MS or noninflammatory neurologic disorders, and IL-6 levels in serum and CSF are elevated during NMOSD relapses. [14, 15, 16] Interleukin-6 promotes the differentiation of naive T cells into proinflammatory type 17 helper T cells, which, along with IL-6, promote the differentiation of B cells into AQP4-IgG–producing plasmablasts.
As previously stated, ON can occasionally result from an infectious process involving the orbits or paranasal sinuses or occur in the course of a systemic viral infection. [20, 21, 22, 23, 24, 25, 26, 27, 28]
Epidemiology
Investigators in Sweden and Denmark have reported an annual incidence of 4 to 5 cases of new-onset optic neuritis (ON) per 100,000 persons. [38] Patients living in temperate climates seem to be predisposed to ON.
Race-, sex-, and age-related demographics
Optic neuritis (ON) appears to affect White individuals more commonly than members of other races. Women are affected twice as often as men. [32]
Typically, patients with first-time, acute ON are young adults 20 to 45 years. Atypical cases of ON may be seen in elderly patients. Bilateral ON in childhood is not uncommon, and it is believed that the risk for progression to MS is lower in childhood.
Prognosis
In contrast to ischemic optic neuropathies and compressive optic neuropathies, optic neuritis (ON) is associated with a gradual recovery of visual acuity with time. [1] For most patients with ON, visual function begins to improve 1 week to several weeks after onset, even without any treatment. However, permanent residual deficits in color vision and contrast and brightness sensitivity are common. [2]
Decreased visual acuity secondary to ON may be permanent. Final visual outcome may be better in patients with an isolated episode of ON, compared with patients who eventually develop multiple sclerosis (MS). Up to 75% of female patients and 35% of male patients initially presenting with ON ultimately develop MS. [39, 40, 41]
Patients with silent demyelinated lesions elsewhere in the brain, observed on magnetic resonance imaging (MRI) performed at the initial presentation, are more likely to develop definite symptomatic MS in the long term than are patients with isolated ON. In addition, patients who have recurrent episodes of ON may be more likely to develop MS.
The 10-year risk of developing clinically definite MS after a single episode of ON was 38% in the entire Optic Neuritis Treatment Trial (ONTT) study group; the 12-year risk was 40%. Most of those who developed MS did so within the first 5 years after the initial episode of ON.
The strongest predictor of MS in the study group was the presence of brain lesions on MRI at the time of the ON episode. Within the study group, patients with at least 1 brain lesion on MRI at the time of the ON episode had a 56% risk of developing symptomatic MS within 10 years. Patients with "normal" MRI findings had a 16% risk for progression to clinically definite MS at 5-year follow-up; this increased to a 22% risk for MS within 10 years.
Most patients with relapsing NMO have an aggressive form of the disease that is associated with frequent and severe exacerbations and poor prognosis.
-
A case of acute optic neuritis. A. 1.5 Tesla, contrast-enhanced spin echo T1-weighted, fat-suppressed coronal MRI through the orbits shows enlargement and contrast enhancement of the left optic nerve in the retrobulbar portion (arrow). B. Coronal spin echo T1-weighted, fat-suppressed MRI of the same patient shows enlargement and contrast enhancement of the nerve in a parasagittal oblique section (arrow).








