Practice Essentials
Neuro-ophthalmic manifestations frequently are encountered in persons with multiple sclerosis (MS) and other central nervous system (CNS) inflammatory disorders, including neuromyelitis optica spectrum disorders (NMOSD) and myelin oligodendrocyte glycoprotein antibody associated disease (MOGAD).
Affected individuals may experience problems with how they see the world (afferent visual pathway symptoms) and/or how smoothly and synchronously their eyes move together (efferent visual pathway disorders). Optic neuritis is an inflammatory injury of the optic nerve that causes vision loss, which is common in MS and other CNS inflammatory disorders. Some individuals with MS and other CNS inflammatory syndromes also may experience homonymous visual field defects caused by lesions in retrochiasmal or retrogeniculate (posterior) regions of the afferent visual pathway. Efferent visual pathway lesions can create a perception of oscillopsia, a visual disturbance in which objects appear to jiggle or move owing to nystagmus (involuntary eye movements). Seeing two objects instead of one (diplopia) with a binocular view can arise from ocular misalignment caused by lesions of the brainstem and cerebellum.
Because patients with MS and other CNS inflammatory disorders who have visual symptoms often seek ophthalmic attention, eye care experts play a vital role in the localization and diagnosis of these conditions.
Special considerations
Patients with visual disturbances in the setting of suspected or established multiple sclerosis (MS) or other CNS inflammatory disorder should be cautioned to avoid activities that present a safety risk to themselves or others. In some instances, this may require a patient to cease driving, at least temporarily.
Patient education
Various resources are available to patients who present with visual manifestations linked to multiple sclerosis (MS) and other CNS inflammatory disorders, as follows:
Research resources are as follows:
-
African School of Neuroimmunology (AfSNI)
Professional organizations are as follows:
Multiple Sclerosis
Multiple Sclerosis (MS) is a CNS disorder that is characterized by both inflammatory and neurodegenerative mechanisms of brain and spinal cord injury. [1, 2] Consequently, most patients with MS experience both episodic relapses and progressive neurologic impairment throughout their disease course. Affecting more than 2 million people worldwide, MS is the leading cause of nontraumatic neurologic disability in young adults. [2] The diagnosis of MS can be established based on clinical and radiologic criteria in patients who experience two or more neurologic events (one of which can be radiologic) consistent with CNS inflammation that are disseminated in space (DIS) and disseminated in time (DIT). Since the publication of the original McDonald criteria [3] and subsequent iterations, [4, 5, 6] radiologic endpoints play an important role in the diagnosis of MS.
Most (85%) patients with MS begin their clinical disease course with episodes of neurologic dysfunction (relapses) followed by complete or incomplete recovery. [1, 2] Over time, many patients with relapsing-remitting MS (RRMS) transition to secondary progressive multiple sclerosis (SPMS). Patients with SPMS present with features of progressive neurologic disability, with or without clinically overt relapses, and insidious disease progression. [1, 2] Approximately 15% of patients with MS experience a primary progressive course from onset, either without preceding relapses (known as primary progressive multiple sclerosis [PPMS]) or with superimposed neurologic events, known as progressive relapsing MS. [1, 2]
Diagnostic criteria for multiple sclerosis
Current diagnostic criteria for MS include clinical and paraclinical measures (Table 1). To render the diagnosis of MS, due diligence is necessary to exclude alternative diagnoses. The revised McDonald criteria [5, 6] rely on the principle that an attack is a neurologic disturbance consistent with the diagnosis of MS persisting for 24 hours or more in the absence of fever or infection. [5, 6] Dissemination in time (DIT) refers to the development or appearance of new CNS lesion over time. [6] Dissemination in space (DIT) refers to the development of lesions in distinct anatomic locations within the CNS. [6]
Table 1. The 2017 McDonald Criteria for Diagnosis of MS in Patients with an Attack at Onset (modified from Thompson et al 2017) [6] (Open Table in a new window)
Number of Clinical Attacks |
Number of Lesions with Objective Clinical Evidence |
Additional Data Required for MS Diagnosis |
≥ 2 |
≥ 2 |
None |
≥ 2 |
1 (plus clear evidence of a previous attack involving a lesion in a distinct neuroanatomic location) |
None |
≥ 2 |
1 |
DIS demonstrated by an additional clinical attack implicating a different CNS location or by MRI a |
1 |
>2 |
DIT demonstrated by an additional clinical attack or by MRI b OR demonstration of CSF OCBs |
1 |
1 |
DIS demonstrated by an additional clinical attack implicating a different CNS site or by MRIa AND DIT shown by an additional clinical attack or by MRIb OR demonstration CSF OCBs |
Abbreviations: CSF = cerebrospinal fluid; CNS = central nervous system; DIS = dissemination in space; DIT = dissemination in time; MRI = magnetic resonance imaging; OCBs = oligoclonal bands
Magnetic Resonance Imaging (MRI) Criteria: aMRI DIS can be indicated by 1 or more T2- hyperintense lesions that are characteristic of MS in 2 or more of the 4 areas of the CNS: periventricular, cortical or juxtacortical, infratentorial, and spinal cord. bMRI DIT can be indicated by the simultaneous presence of gadolinium-enhancing and nonenhancing lesions at any time or by a new T2-hyperintense or gadolinium-enhancing lesion on follow-up MRI, with reference to a baseline scan, regardless of the timing of the baseline scan. |
||
Approved disease-modifying therapies for multiple sclerosis
Disease-modifying therapies (DMTs) used to treat multiple sclerosis (MS) decrease clinical and subclinical central nervous system (CNS) inflammation with the goal of reducing accumulated disability and disease progression. These medications are used to treat patients with relapsing-remitting MS (RRMS), patients with a clinically isolated syndrome (CIS) who are at risk of developing MS, and patients with secondary progressive MS (SPMS). Because of the chronic nature of MS and the potential adverse effects of therapy, treatment must be tailored to the needs of the patient, ideally by a clinician with expertise in managing the disease.
Table 2. Approved Disease-Modifying Therapies (DMTs) Used in the Treatment of Multiple Sclerosis [7, 8, 9, 10, 11, 12, 13] (Open Table in a new window)
Drug |
Route |
MS Subtype |
Dosing Frequency |
Most Common Adverse Effects |
Interferon β-1a (Avonex) |
IM |
Relapsing forms of MS, CIS |
Once weekly |
Flulike symptoms Liver enzyme changes BMS Thyroid dysfunction |
Interferon β-1a (Rebif) |
SC |
Relapsing forms of MS, CIS |
22 μg or 44 μg 3 times weekly |
Flulike symptoms Liver enzyme changes BMS Thyroid dysfunction |
Interferon β-1b (Betaseron) (Extavia) |
SC |
Relapsing forms of MS, CIS |
3 times weekly |
Flulike symptoms Liver enzyme changes BMS Thyroid dysfunction |
Glatiramer acetate (Copaxone) Glatopa (glatiramer acetate–generic equivalent of Copaxone 20mg and 40 mg doses) |
SC |
Relapsing forms of MS, CIS |
Once daily |
Skin irritation Skin lipoatrophy Panic attack–like events (often self limited) |
| Peginterferon beta 1a (Plegridy) | SC or IM | Relapsing forms of MS, CIS | Every 2 weeks | Flulike symptoms Liver enzyme changes Depression/suicide Injection site reactions Low peripheral blood counts CHF Seizures Thrombotic microangiopathy Autoimmune disorders |
Natalizumab (Tysabri) |
IV |
Relapsing forms of MS, CIS |
Once monthly |
Nausea Infection Liver dysfunction Binding antibodies PMLa |
Fingolimod (Gilenya) |
PO |
Relapsing forms of MS, CIS |
Once daily |
Macular edema Bradyarrhythmia QT interval prolongation HTN Severe varicella-associated complications in nonimmune patients Increased risk for HZ in all patients Mild infections PML |
Dimethyl fumarate (Tecfidera) |
PO |
Relapsing forms of MS, CIS |
Twice daily |
Flushing GI distress Lymphopenia PML (rare) |
| Monomethyl fumarate (Bafiertam) | PO | Relapsing forms of MS, CIS | Twice daily | Flushing GI illness HZ, opportunistic infections Lymphopenia PML (rare) |
Teriflunomide (Aubagio) |
PO |
Relapsing forms of MS, CIS |
Once daily |
Nausea Headaches Alopecia Liver dysfunction Presumed teratogenicity |
Alemtuzumab (Lemtrada) |
IV | Relapsing forms of MS |
Minimum of 2 cycles (baseline and Year 1) |
Infusion reactions Mild to moderate infections Thyroid dysfunction ITP Anti–GBM disease Stroke Alveolar hemorrhage Autoimmune hemolytic anemia Hemophagocytic lymphohistiocytosis |
Ocrelizumab (Ocrevus) |
IV |
Relapsing forms of MS, CIS |
Every 6 months |
Hypogammaglobulinemia Infections (skin, urinary, sinus) Infusion reactions Rare neutropenia and lymphopenia Uncertain if true link to breast cancer |
Ofatumumab (Kesimpta) |
SC |
Relapsing forms of MS, CIS |
Once monthly |
Injection site reactions URIs UTIs Headache Reduced IgM |
Cladribine (Mavenclad) |
PO | Relapsing forms of MS |
Yearly (for 2 years) |
Headaches Lymphopenia HZ infections Nausea Abdominal pain Bronchitis Diarrhea Increased risk for routine infections |
Siponimod (Mayzent) |
PO |
Relapsing forms of MS, CIS |
Once daily |
Lymphopenia Increased LFTs Rare bradycardia Macular edema HTN Varicella zoster infection |
Ozanimod (Zeposia) |
PO |
Relapsing forms of MS, CIS |
Once daily |
URIs Hepatic transaminase elevations Orthostatic hypotension UTIs Back pain HTN |
| Ponesimod (Ponvory) | PO | Relapsing forms of MS, CIS | Once daily | Infections Bradyarrhythmia Liver enzyme abnormalities HTN |
| Diroximel fumarate (Vumerity) | PO | Relapsing forms of MS, CIS | Twice daily | Flushing GI illness HZ, opportunistic infections Lymphopenia PML (rare) |
Abbreviations: CIS = clinically isolated syndrome; MS = multiple sclerosis; GI = gastrointestinal; HZ = herpes zoster; CHF = congestive heart failure; HTN = hypertension; URI = upper respiratory tract infection; UTI = urinary tract infection; BMS = bone marrow suppression; ITP = Idiopathic thrombocytopenic purpura; Anti-GBM disease = anti–glomerular basement membrane disease; LFT = liver function test; PML = progressive multifocal leukoencephalopathy. aRisk of PML risk increases with a history of immunosuppression, JC virus positivity, and use of the drug for more than 2 years. |
||||
Neuromyelitis optica spectrum disorders (NMOSD)
Neuromyelitis optica spectrum disorders(NMOSD) refers to a group of CNS inflammatory conditions distinct from multiple sclerosis (MS). [14] Our understanding of NMOSD has rapidly evolved (it is autoimmune astrocytopathy) with the introduction of a serologic marker and adaptation of revised diagnostic criteria. [14] [15] In 2004, detectable serum antibodies targeting the water channel aquaporin-4 (AQP4–IgG) were discovered. [16, 17] In 2015, the International Panel for NMO Diagnosis was convened to revise diagnostic criteria. [14]
From a clinical perspective NMOSD tends to affect middle aged women (mean age of optic neuritis onset is 40 years, and women can be affected nine times as frequently as men). [17] NMOSD is more common in individuals or Asian or Afro-Caribbean descent. The majority of patients with NMOSD have a relapsing course. [17]
New nomenclature was defined, including the unifying term NMOSD, which is further stratified by serologic testing (NMOSD with or without AQP4-IgG). [14] The core clinical characteristics required for a diagnosis of antibody-positive NMOSD include clinical syndromes or magnetic resonance imaging (MRI) findings related to optic nerve, spinal cord, area postrema, other brainstem, diencephalic, or cerebral presentations. [14] Notably, robust clinical criteria with additional neuroimaging findings are required for diagnosis of NMOSD without proof of AQP4-IgG positivity. [14]
Table 3. NMOSD Diagnostic Criteria for Adult Patients [14] (Open Table in a new window)
|
NMOSD with + AQP4-IgG
|
NMOSD with AQP4-IgG Negative/Unknown Status
At least 2 core clinical characteristics resulting from 1 or more clinical attacks, meeting the following requirements:
|
Core Clinical Characteristics
|
a Additional MRI Requirements
|
| Abbreviations: AQP4-IgG = aquaporin 4–IgG ; MRI = magnetic resonance imaging; NMOSD = neuromyelitis optica spectrum disorders |
The diagnosis of NMOSD is best confirmed by cell-based serum assays, which are approximately 80% sensitive for NMOSD diagnosis. [14] [17] In addition to MRI, cerebrospinal fluid (CSF) analysis can help facilitate the diagnosis. The absence of CSF oligoclonal bands (OCBs) is considered supportive evidence for NMOSD in the appropriate clinical context. Moreover, CSF pleocytosis—often including elevated neutrophils and/or eosinophils—is associated with a higher risk for NMOSD. [14, 17]
Patients meeting the diagnostic criteria for NMOSD generally are treated with immunosuppressive agents, as opposed to disease-modifying therapies used in the treatment of patients with MS, because the latter can be harmful. [17] Immunosuppressive therapy should be initiated immediately, often in concert with acute high-dose corticosteroid therapy in the context of a relapse, to prevent further neurologic disability for patients with NMOSD.
The optimal first-line agent and duration of treatment in this context are uncertain. Historically, rituximab, azathioprine, mycophenolate mofetil, and long-term corticosteroid therapy have been prescribed. [17, 18] Phase 3 placebo-controlled clinical trials have demonstrated the efficacy and safety of 3 new therapeutics that significantly reduce the risk of future NMOSD attacks including eculizumab, satralizumab, and inebilizumab. [17, 18, 19, 20, 21, 22]
Myelin oligodendrocyte glycoprotein IgG–associated disease
Myelin oligodendrocyte glycoprotein antibody-associated disease (MOGAD) refers to another spectrum of inflammatory demyelinating central nervous system (CNS) syndromes characterized as being the target of myelin oligodendrocyte glycoprotein (MOG) IgG1 antibodies. A MOGAD syndrome may have a monophasic or relapsing course of neurologic dysfunction, which does not meet the typical criteria for multiple sclerosis (MS) or other related CNS inflammatory disorders, and it occurs in association with serum MOG antibodies. [23]
MOGAD has been associated with demyelinating N-methyl-d-aspartate receptor encephalitis, postinfectious demyelination following herpes simplex virus, Borrelia and Epstein-Barr virus infections, and rarely, relapsing MS. [23] It is unclear whether MOG antibodies play a pathogenic role in all these conditions or whether the relationship is merely a bystander effect. [23]
MOGAD is much more gender balanced than NMOSD and is characterized by a median age at onset in the early to middle thirties. [23] Notably, optic neuritis is the most common heralding event for 50-60% of affected individuals, followed by myelitis, acute disseminated encephalomyelitis (ADEM), or an ADEM-like presentation (eg, brainstem attacks), which are especially common in children. [23] A relapsing course has been reported in a wide range of patients with MOGAD (44-83%) and commonly involves the optic nerve. [23]
Most patients with MOGAD also have CNS lesions detected on baseline magnetic resonance imaging (MRI). The majority of MRI lesions are bilateral, and approximately one-third are subtentorial, often affecting the brainstem. [23] Thalamic and pontine lesions are more common in MOGAD than in seropositive neuromyelitis optica spectrum disorders (NMOSDs). [23]
In children, bilateral thalamic lesions at onset are frequently observed, affecting approximately 60% of patients. [23] More than half of patients with MOGAD demonstrate T2 hyperintense lesions in the spinal cord; these lesions are relatively short and visible in the cervical or thoracic region, [23] and they may also involve the conus. It is noteworthy that cerebrospinal fluid (CSF) pleocytosis occurs in a large number of patients with MOGAD (44-85%) and is more commonly observed in children. [23] As in patients with NMOSD, positive oligoclonal bands (OBCs) are unusual in the cerebrospinal fluid (CSF), occurring in only 6% of patients with MOGAD. [23] Furthermore, CSF protein is elevated in around a third of patients with MOGAD. [23]
The diagnosis of MOGAD is best facilitated when MOG IgG antibodies are detected in serum by means of a cell-based assay. [23, 24] MOG antibody titers can be higher during relapses relative to periods of remission and in patients with a relapsing MOGAD disease course. In children, persistent high-titer MOG antibodies have been associated with a risk for relapse. [23]
High-dose intravenous methylprednisolone is often used for the treatment of MOGAD manifestations (dose ranging from 1 to 2 g once a day for 3-5 days). [23] Plasma exchange (regimens varying from 3 to 5 cycles) and intravenous immunoglobulin IgG also may be used. {ref 23}The optimal therapy and duration of treatment remain unclear because data from randomized controlled trials are lacking.
Glial fibrillary acidic protein IgG–associated disease
In 2016, an antibody to glial fibrillary acidic protein (GFAP-IgG) in cerebrospinal fluid (CSF) was found to be specific for an inflammatory meningoencephalomyelitis, termed autoimmune GFAP astrocytopathy. [24] Clinical manifestations of GFAP IgG-associated disease include subacute to chronic meningitis, optic disc swelling, encephalitis, and myelitis. [24] Male and female individuals can be affected equally with age at onset usually in the mid-forties. [24] Coexisting neoplasms (particularly teratoma) have been associated with GFAP IgG antibody-associated disease. [24]
Unlike testing for neuromyelitis optica spectrum disorders (NMOSD) and myelin oligodendrocyte glycoprotein IgG–associated disease (MOGAD), testing for GFAP IgG antibodies is best performed with cerebrospinal fluid (CSF) in lieu of serum for optimal sensitivity and specificity. [24] In patients with GFAP IgG–associated disease, cerebral magnetic resonance imaging (MRI) may reveal a characteristic radial pattern of perivascular enhancement perpendicular to the ventricles. [24] Leptomeningeal enhancement also is commonly observed. [24]
In some patients with GFAP IgG–associated disease, a longitudinally extensive T2 hyperintensity signal or punctate parenchymal enhancement may be seen in the spinal cord. [24] Analysis of CSF obtained from these patients often demonstrates an elevated white blood cell count; in some cases, CSF oligoclonal bands (OCBs) may be observed. [24] High dose corticosteroids are the mainstay of therapy. Corticosteroid-sparing agents are used to try to maintain remission of GFAP IgG–associated disease, although randomized controlled trial data are lacking. [24]
Table 4. Clinical, Serologic, And Radiologic Features Distinguishing MS, NMOSD, and MOGAD [6, 10, 14, 17, 18, 20, 24, 25] (Open Table in a new window)
Feature |
MS |
NMOSD |
MOGAD |
Age |
Variable (median age at onset in 3rd decade) |
Variable (median age at onset > age at onset of MS and in 4th decade) |
Variable |
Female: Male |
2-3:1 |
9:1 |
1.5:1 |
Epidemiology |
White persons more often affected |
Nonwhite persons including Black persons and persons of Asian and Afro-Caribbean descent more predisposed |
Unknown |
Serologic Marker |
None |
AQP4-IgG |
MOG-IgG |
Brain MRI |
Ovoid periventricular, juxtacortical, cortical, infratentorial peripheral, ring-/open ring-enhancing T2, FLAIR, Gd lesions |
Can be normal or show nonspecific lesions; classic lesions may also be found in the area postrema, region of the 3rd/4th ventricle, splenium of the corpus callosum (20% of patients may have MS-appearing lesions) |
Lesions that are ADEM-like, involving the deep gray matter, appearing diffuse/ confluent in the brainstem, affecting cerebellar peduncles |
Optic Nerve MRI |
Often unilateral, involving short segments (< 50%) of affected nerve length |
Often bilateral with enhancement of >50% of optic nerve, particularly involving posterior regions of the anterior visual pathway including chiasm |
Often bilateral with enhancement of >50% of optic nerve, particularly intraorbital optic nerve with perineural involvement |
Spine MRI |
Multiple lesions may be seen including short lesions; periphery of cord (dorsal/lateral column); ring or variable enhancement |
Single lesion (LETM) and short areas of segmental involvement may be seen with variable enhancement |
Multiple lesions may be seen (75% LETM; 25% short); the conus may be involved and lesional enhancement is variable |
Acute Treatment |
High-dose corticosteroids (rarely plasma exchange) |
High-dose corticosteroids ± plasma exchange |
High-dose corticosteroids ± plasma exchange/IVIG (in children) |
Long-term Management |
DMTs |
Azathioprine, mycophenolate mofetil, rituximab; newer targeted agents include eculizumab, satralizumab, and inebilizumab |
Options for recurrent disease include azathioprine, mycophenolate mofetil, rituximab |
|
|||
Abbreviations: ADEM = acute disseminated encephalomyelitis; AQP4 = aquaporin 4; DMTs = disease-modifying therapies; FLAIR = fluid-attenuated inversion recovery; Gd = Gadolinium; LETM = longitudinally extensive transverse myelitis; MOG = myelin oligodendrocyte glycoprotein; MOGAD = myelin oligodendrocyte glycoprotein antibody–associated disease; MRI = magnetic resonance imaging; MS = multiple sclerosis; NMSOD = neuromyelitis optica spectrum disorders
Afferent Visual Pathway Manifestations of Multiple Sclerosis and Related Disorders
Optic neuritis is an inflammatory injury of the optic nerve that represents the best characterized CIS associated with MS. [26, 27] In fact, approximately 20% of patients with MS present with optic neuritis as the first clinical disease manifestation. [26] It is important to note that the vast majority of optic neuritis cases encountered in clinical are either sporadic or MS related. At a population level, optic neuritis associated with NMOSD and MOGAD account for 9% of optic neuritis cases. [28]
Clinical features at presentation
Many of the cardinal clinical features of typical optic neuritis associated with MS have been identified based on the Optic Neuritis Treatment Trial (ONTT). [29, 30] The ONTT revealed that most patients with typical optic neuritis are White (85%) and female (77%) with a mean age of approximately 32 years. [29, 30]
Among adults, sporadic optic neuritis cases are typically unilateral; however, bilateral simultaneous vision loss may be observed. Nonetheless, in this setting, other demyelinating optic neuropathies and potential optic neuritis mimics must be considered, including NMOSD, MOGAD, and Leber hereditary optic neuropathy (LHON) (see Table 5).
From a clinical perspective, a comprehensive history is imperative for differentiating optic neuritis from its potential mimics. Patients with optic neuritis often report subacute-onset vision loss that worsens over hours to days. This mode of onset helps distinguish optic neuritis from non-arteritic anterior ischemic optic neuropathy (NA-AION), for example. [31] In contrast, NA-AION generally is unaccompanied by pain, and symptoms frequently are noted upon morning awakening. Patients with compressive optic neuropathies may report sudden-onset awareness of vision loss. However, a detailed discussion often reveals that these individuals have experienced sudden-onset awareness of a longer-standing problem, rather than sudden-onset vision loss itself. [31]
Ninety-two percent of individuals with typical optic neuritis experience pain that is frequently provoked by eye movements, within the first 2 weeks of symptom onset. [31] Patients also may report intermittent flashes of light in the affected eye, known as photopsias or phosphenes. [32] Patients with optic neuritis may also describe worsening vision with increased body temperature, referred to as Uhthoff phenomenon. [31, 33]
Common examination findings in patients with optic neuritis
In patients with suspected optic neuritis, several features on initial examination can help verify the diagnosis. Initially, the severity of vision loss in the affected eye may range from mild (Snellen visual acuity equivalent of 20/20) to—in rare cases—no light perception with high-contrast letter acuity testing. [31] In patients with unilateral optic neuritis or bilateral optic neuritis with asymmetric involvement, a relative afferent pupillary defect (RAPD) is apparent in the affected or, in cases of bilateral involvement, the more severely affected eye. [31] Visual field loss in patients with optic neuritis tends to follow the topography of the retinal nerve fiber layer (RNFL), with cecocentral, altitudinal, and arcuate deficits frequently observed. [31, 34, 35]
Keltner and colleagues [34] classified visual field abnormalities observed during longitudinal follow-up of patients in the ONTT and reported that both the affected and fellow eyes in patients with optic neuritis showed visual field losses. [34] These findings illustrate the role of perimetry in detecting both clinically overt and clinically occult optic nerve involvement in patients with MS.
Dyschromatopsia, or decreased color vision, is typical in eyes with optic neuritis. [31] This finding can be particularly helpful in confirming the diagnosis in patients with mild central vision loss who have disproportionate deficits in color vision function. [31] Often, patients continue to note subjective color desaturation in the affected eyes after high-contrast visual acuity function has returned to a Snellen equivalent of 20/20.
Traditionally, in typical cases of retrobulbar optic neuritis, the optic nerve has been described as normal in appearance, whereas patients with anterior optic neuritis, or papillitis, present with mild to moderate optic disc swelling at presentation. [31] As exemplified in the ONTT, severe optic disc edema, [29, 30, 31] vitreous cells, and/or hemorrhage are uncommon in the setting of typical optic neuritis. Therefore, the observation of these fundus features should prompt investigation for other potential causes of vision loss.
Pediatric optic neuritis
Several features of the clinical presentation and course differentiate pediatric optic neuritis from the adult-onset syndrome. While both children and adults present with vision loss, pain upon eye movements, dyschromatopsia, and visual field defects, children are more likely to present with more severe vision loss.
Children with optic neuritis more frequently demonstrate papillitis or anterior optic neuritis than their adult counterparts. Moreover, younger pediatric patients are more likely than adolescents or adults to experience bilateral simultaneous optic nerve involvement. In retrospect, the phenotypic characteristics that have long distinguished pediatric optic neuritis cases from adult version of optic neuritis have in fact reflected that many of these cases in children were part of the MOGAD spectrum. Notably, MOG antibodies are more commonly encountered than AQP4-IgG antibodies in pediatric patients. [36, 37] In fact, among children with optic neuritis, 50% will have MOG antibodies. [37] As mentioned, involvement of the anterior optic pathway (papillitis) is more frequently seen with MOGAD as compared to NMOSD patients who more often have lesions involving the posterior pathway with both chiasmal and post chiasmal involvement. [37] In both NMOSD and MOGAD bilateral optic neuritis and the presence of longitudinal lesions in the anterior visual pathway are distinguishing features. [37]
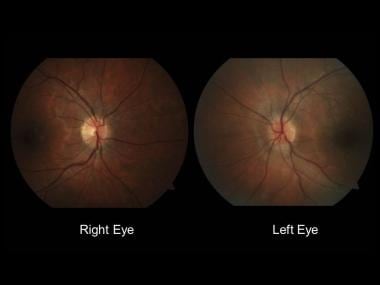
Optic neuritis: Making the diagnosis
In patients with a typical history and expected examination findings, optic neuritis can be reliably diagnosed based on clinical grounds; however, certain red flags should raise concern for a potential mimic and therefore prompt additional investigations.
For example, young men who present with painless bilateral sequential or simultaneous optic nerve dysfunction should undergo testing for LHON. Middle-aged women in whom unilateral or bilateral optic neuritis develops should be evaluated for NMOSD and MOGAD because the management of optic neuritis in patients with these CNS inflammatory disorders differs from that of patients with MS. Additional features that should increase the clinical suspicion for NMOSD include poor clinical recovery, lack of typical MRI findings for MS, cerebrospinal fluid (CSF) pleocytosis, and manifestations of transverse myelitis. As described in the sections to follow, some patients with recurrent optic neuritis harbor a diagnosis of NMOSD or MOGAD.
Table 5. Differential Diagnoses of Optic Neuritis (Modified from Table 2-1 in Costello F. Inflammatory optic neuropathies. Continuum (Minneap Minn). Aug 2014; 20 (4 Neuro-ophthalmology): 816-37.] [28] (Open Table in a new window)
Diagnosis |
Clinical Features |
Investigations to Consider |
NA-AION |
Painless, altitudinal visual field defect common; vision loss on awakening; vascular risk factors include phosphodiesterase type 5 inhibitor use, nocturnal antihypertensive use, sleep apnea; physiologic disc at risk, patients with NA-ION have optic disc edema acutely |
Sleep study, 24h BP monitoring, investigations for HTN and diabetes |
Compressive optic neuropathy (pituitary lesions, meningiomas, aneurysm) |
Painless, progressive vision loss; color loss disproportionate to visual acuity deficit; non-glaucomatous optic disc cupping; temporal visual field cut; bilateral visual field involvement |
Cranial and orbital MRI/MRA or CT/CTA |
Infectious optic neuropathies (eg, tuberculosis, syphilis, Lyme disease) |
Associated uveitis, papillitis, or retrobulbar optic neuropathy; macular star; infectious symptomatology |
Serum/CSF culture/sensitivity; specific serologic testing for syphilis, Lyme, Bartonella henselae, HIV, toxoplasmosis, HBV and HCV; EBV; histoplasmosis; TB testing; chest imaging; ESR, CRP |
Inflammatory/demyelinating optic neuropathies not associated with MS or an underlying systemic disorder: NMOSD, CRION, ADEM, and MOGAD |
Poor recovery, unilateral or bilateral optic neuritis, associated transverse myelitis, recurrent symptoms |
Brain MRI, cervical spine MRI, serum anti-AQP4 IgG antibody testing, serum anti-MOG IgG antibody testing |
Genetic optic neuropathies (LHON, autosomal-dominant optic neuropathy) |
Bilateral vision loss, painless, poor recovery, family history |
Genetics referral with specific mutation testing |
Toxic/nutritional (tobacco-alcohol amblyopia and Cuban and Tanzanian epidemic optic neuropathies) |
Bilateral optic nerve involvement, history of drug use (ethambutol, selenium, amiodarone), restricted nutritional intake, glue sniffing, methanol ingestion |
Vitamin B12 levels, toxicology screen |
Sarcoid optic neuropathy |
Steroid responsive, poor recovery, systemic symptoms and signs |
Chest imaging, serum ACE, gallium scan, tissue diagnosis, bronchoalveolar lavage, soluble IL-2 receptor |
Connective tissue/vasculitic optic neuropathy (lupus, Wegener granulomatosis, Sjögren syndrome, Behçet disease) |
Steroid responsive, associated systemic symptoms and signs |
Serum ESR, Sjögren-specific antibodies, CRP, ANCA, ENA panel, ANA |
Orbital inflammation/optic perineuritis |
Orbital signs (proptosis) |
MRI or CT orbital imaging, blood work including TSH ANCA, CRP, ESR, ACE |
Uveitis/posterior scleritis |
Severe pain, floaters, vitreous reaction |
Fluorescein angiography, B-scan ultrasonography of orbits |
Autoimmune optic neuropathy (similar to CRION) |
Steroid responsive |
Skin biopsy for immunoglobulin deposition |
Big blind spot syndromes |
Blind spot on visual field testing, painless photopsias, bilateral ocular involvement |
Full-field/multifocal ERG, fluorescein angiography |
Abbreviations: ACE = angiotensin-converting enzyme; ADEM = acute disseminated encephalomyelitis; anti-DS DNA = anti-double-stranded DNA; anti-MOG = anti–myelin oligodendrocyte glycoprotein; ANA = antinuclear antibody; ANCA = antineutrophilic cytoplasmic antibody; AON = autoimmune optic neuropathy; AQP4 = aquaporin 4; CRION = chronic relapsing inflammatory optic neuropathy; CRP = C-reactive protein; CSF = cerebrospinal fluid; CT = computed tomography; CTA = computed tomographic angiography; ENA = extractable nuclear antigen; ERG = electroretinogram; ESR = erythrocyte sedimentation rate; FTA-ABS = fluorescent treponemal antibody absorption; LHON = Leber hereditary optic neuropathy; MOG = myelin oligodendrocyte glycoprotein; MOGAD = myelin oligodendrocyte glycoprotein IgG-associated disease; MRA = magnetic resonance angiography; MRI = magnetic resonance imaging; MS = multiple sclerosis; NA-ION = nonarteritic anterior ischemic optic neuropathy; NMO-IgG = neuromyelitis optica IgG; NMSOD = neuromyelitis optica spectrum disorder; PCR = polymerase chain reaction; TSH = thyroid-stimulating hormone; VDRL = Venereal Disease Research Laboratory; ; HBV = hepatitis B virus; HCV = hepatitis C virus; EBV = Epstein-Barr virus; ESR = serum sedimentation rate; CRP = C-reactive protein |
||
Optic neuritis: Ancillary studies
In the setting of typical optic neuritis, laboratory studies are not generally useful in facilitating diagnosis [32] ; however, additional investigations can be helpful in detecting potential mimics, such as NMOSD, MOGAD, lupus-related optic neuropathy, and syphilitic optic nerve injury.
Specific clinical features should prompt consideration of these alternate diagnoses. Therefore, ancillary investigations should be selected based on the history and physical examination findings and may include any of the following:
-
Complete blood count (CBC) to evaluate for features of anemia, leukemia, or leukocytosis
-
Serum vitamin B12 and folate levels (eg, bilateral central scotoma)
-
Lyme titers (eg, endemic area, tick exposure, rash of erythema chronica migrans)
-
Tuberculin skin testing, chest radiography, or QuantiFERON-TB testing (eg, tuberculosis [TB] exposure, endemic area)
-
Fluorescent treponemal antibody (FTA) testing (eg, syphilis serology) or nontreponemal testing (eg, Venereal Disease Research Laboratories [VDRL] testing or rapid plasma reagin [RPR] testing)
-
Antinuclear antibody (eg, systemic lupus erythematosus)
-
HIV testing (eg, high-risk patients)
-
Angiotensin-converting enzyme (ACE) level, lysozyme (eg, sarcoidosis)
-
Erythrocyte sedimentation rate (eg, inflammatory disorders)
-
Serum NMOSD antibody IgG (anti–aquaporin-4 [AQP4] antibody) testing
-
Serum MOG IgG antibody testing
-
Anti-GAD 65 antibodies
-
Mononuclear spot test (Monospot test) (infectious mononucleosis due to Epstein-Barr virus)
-
Cerebrospinal fluid analysis
-
Computed tomography scan of chest/abdomen/pelvis in the appropriate setting (sarcoidosis, lymphoma, other malignancies)
-
Whole-body positron emission tomography (PET) or computed tomography (CT) in the appropriate setting (sarcoidosis, lymphoma, other malignancies)
Cerebrospinal fluid analysis generally is not required to diagnose typical cases of optic neuritis. The presence of oligoclonal bands (OCBs) can assist with the diagnosis of MS in patients who meet dissemination in space criteria, but not dissemination in time criteria. Cerebrospinal fluid analysis can also assist with determining the presence of alternative entities, such as suspected infectious optic neuropathies secondary to syphilis, tuberculosis, or Lyme disease.
Visual evoked potentials
In clinical practice, visual evoked potential (VEP) testing typically is unnecessary to confirm the diagnosis of optic neuritis. When mild optic neuritis or subclinical optic nerve damage is suspected, VEP testing can be useful in capturing the effects of prior demyelinating injury. [2] Abnormal VEP findings in this context include increased latencies and reduced amplitudes of waveform. [2] However, VEP abnormalities are not restricted to optic neuritis and may also occur with other conditions, such as optic nerve compression, infiltration, and nondemyelinating inflammation. [38] Multifocal VEP can be a more sensitive and specific tool for detecting optic neuritis in suspected clinically overt or clinically occult cases of optic neuritis, although the technique is not widely available for routine clinical use. [2] [38]
Optical coherence tomography
Optical coherence tomography (OCT) is a noninvasive ocular imaging technique that provides high-resolution images of retinal architecture in vivo.
Changes in peripapillary retinal nerve fiber layer (RNFL) thickness represent axonal damage, whereas loss of macular volume and ganglion layer thickness provides an indirect measure of neuronal injury in the afferent visual pathway. [39] In the context of acute optic neuritis, OCT-measured peripapillary RNFL thickness tends to be elevated in the eye affected by optic neuritis initially, presumably because of axoplasmic flow stasis. [39] In contrast, macular volume and ganglion layer measures, as determined with OCT, are comparable between affected and unaffected eyes of patients at symptom onset but later decline for up to 12 months. [39]
Visual outcomes after acute optic neuritis—including high- and low-contrast letter acuity, color vision, and visual field sensitivity—correlate with the amount of OCT-measured RNFL, ganglion layer, and macular volume loss detected 6 to 12 months after onset of optic neuritis. [39]
Recurrent optic neuritis, such as that seen with NMOSD, has been associated with poorer OCT measures. In a study of pediatric patients with CNS demyelinating syndromes, there was a 9-μm (9%) decrement in RNFL thickness for each additional optic neuritis episode. [40]
One disadvantage of OCT is that a so-called floor effect can complicate detection of new changes in RNFL thickness in the setting of pre-existing optic atrophy, since mean RNFL values do not decrease below a measure of approximately 30 μm, regardless of the extent of optic nerve injury. [39]
Optic neuritis: Acute management
The Optic Neuritis Treatment Trial (ONTT) showed that high-dose intravenous corticosteroids (250 mg administered every 6 hours for 3 days, followed by oral prednisone [1 mg/kg/day] for 11 days) accelerated visual recovery relative to oral prednisone (1 mg/kg/day) and oral placebo. [29, 30, 31] In the ONTT, the only benefit of corticosteroids was hastened visual recovery within the first 2 weeks, which is the primary indication for treatment. [18] Secondary analyses of the trial data suggest that this early benefit represents only about 1 or 2 lines of Snellen acuity. [18]
Subsequent studies have shown that the bioavailability of 1250 mg of oral prednisone (or 500 mg, administered orally twice daily) is comparable to that of 1 g of intravenous methylprednisolone (IVMP). [41, 42]
Martinelli and colleagues compared the efficacy and safety of 1000 mg of IVMP and 1000 mg of oral methylprednisolone in patients experiencing MS relapse. [41] Both treatment groups demonstrated reduced gadolinium-enhancing MRI lesions over time with a noninferiority effect evident between the 2 routes of administration. Patients in both treatment groups also showed significant improvement in Expanded Disability Status Scale (EDSS) scores in this study. [41]
Burton and colleagues compared the efficacy of oral and intravenous steroids for MS relapses and reported no significant differences in clinical, radiologic, or pharmacologic outcomes between the groups. [42] Therefore, in clinical practice, high-dose oral steroids often are substituted for intravenous treatment because they are a more convenient option for patients and their caregivers.
Patient-related factors should be considered when a decision about whether to initiate or defer treatment with steroids is being made for a patient with optic neuritis. In 2000, the Quality Standards Subcommittee of the American Academy of Neurology (AAN) reviewed the role of high-dose corticosteroids in the treatment of acute optic neuritis. The recommendation from the AAN was that treatment should be given with the intention to hasten recovery but not to improve ultimate visual outcome. Moreover, treatment decisions should take other non–evidence-based factors into account, such as quality of life, risk to the patient, and visual function in the fellow eye. [43]
In contrast to typical optic neuritis associated with MS, optic neuritis associated with NMOSD and MOGAD needs to be more aggressively managed, often with more prolonged steroid regimens.
Prognosis for visual recovery
Visual recovery after typical optic neuritis (associated with MS or sporadic in nature) tends to be favorable and frequently occurs within 3 to 6 weeks of symptom onset. During this time, patients experience optic atrophy, with temporal optic disc pallor. In the ONTT, mean visual acuity 1 year after entry improved to 20/20 (Snellen equivalent), with less than 10% of patients having a visual acuity worse than 20/40. [44] Early features that may predict a less favorable recovery after optic neuritis include visual acuity of 20/50 or worse, contrast sensitivity less than 1.0 log units, and visual field mean deviation of -15 decibels or less 1 month after initial presentation. [45]
Motion perception deficits have been shown to persist a year after optic neuritis, despite recovery of high- and low-contrast visual acuity, color vision, and visual field performance. [46] Persistent deficits in motion perception may partly explain why many patients with optic neuritis describe problems with visual tracking in the postacute phase.
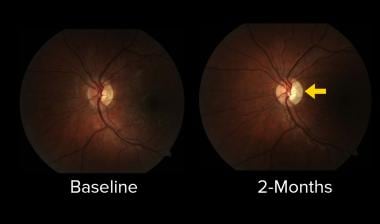
The risk of developing multiple sclerosis after optic neuritis
Studies have shown that the risk of developing MS increases over time in patients who present with optic neuritis as a clinically isolated syndrome (CIS). In a study conducted by Rodriguez and colleagues, [47] the 10-year risk for clinically definite MS was 39%, the 20-year risk was 49%, and the 40-year risk was 60%. In the longitudinal follow-up from the ONTT, the 15-year risk of developing clinically definite MS was 25% in patients with no brain lesions on baseline MRI, compared with 72% in patients with 1 or more lesions. [48]
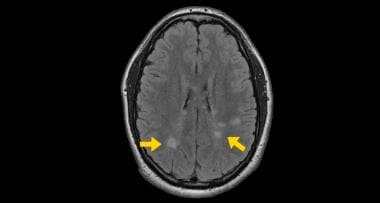
The risk for MS was 3 times higher in women in the ONTT. [48] Moreover, MS was more than twice as likely to develop in patients with retrobulbar optic neuritis as in patients with papillitis. [48] However, in the ONTT era, MS was diagnosed based on clinical criteria, whereas MS can be diagnosed at the time of the first clinical event based on MRI evidence of dissemination of lesions in space and time.
Optic neuritis associated with neuromyelitis optica spectrum disorders and myelin oligodendrocyte glycoprotein antibody–associated disease
In cases of neuromyelitis optica spectrum disorder (NMOSD), optic neuritis is often more bilateral and severe, with visual acuity at presentation being the Snellen equivalent of 20/200 or worse. [14] Altitudinal visual field defects are more likely to be observed, and recovery may be poor despite high-dose corticosteroid therapy. [14] During optic neuritis attacks, lesions within the optic nerves may be detected with fat-suppressed T2-weighted orbital magnetic resonance imaging (MRI) sequences.
Typically, gadolinium enhancement of the optic nerve is seen on T1-weighted sequences. Bilateral optic nerve involvement, with a posterior nerve predominance (especially with extension into the optic chiasm), or extensive lesions of the optic nerve (more than half of the nerve length involved) are all radiologic features suggestive of NMOSD. [14]
Optic neuritis associated with MOGAD typically occurs with or without other neurologic symptoms, and it is often recurrent. Patients often report pain at symptom onset and manifest significant optic disc edema, which distinguishes these cases from optic neuritis associated with multiple sclerosis (MS). [25]
The MRI features of MOGAD-associated optic neuritis include longitudinally enhancing intraorbital lesions of the optic nerve, seen best with T1-weighted fat-suppressed gadolinium-enhanced views. Fifty percent of patients with MOGAD optic neuritis will also manifest perineural involvement with contrast enhancement of the optic nerve sheath and surrounding orbital tissue. [25] Although MOGAD optic neuritis attacks tend to present with severe vision loss, the majority of patients show good clinical recovery, [25] which is different from NMOSD-associated optic neuritis. [17]
Uveitis
Patients with MS are known to experience various forms of ocular inflammation, including uveitis, retinal perivascular sheathing (periphlebitis), and retinitis. [49, 50, 51] Uveitis refers to inflammation of the uveal tract and is 10 times more common in patients with MS (incidence of 1-2%) than in the general population. [52] Intermediate uveitis, or pars planitis, is the most frequent form of uveitis seen in patients with MS.
Patients with uveitis report blurred vision, floaters, photophobia, pain, and eye redness. [52] Ocular complications include retinal neovascularization, cystoid macular edema, cataracts, retinal detachment, and epiretinal membrane formation. [52] Although uveitis and complications thereof can occur as part of MS itself, they may also arise as a consequence of MS therapy. Specifically, patients with MS who are treated with fingolimod, a sphingosine-1-phosphate receptor modulator, may develop fingolimod-associated macular edema (FAME). [51]
The clinical features of FAME typically manifest within months of initiation of therapy, although patients may or may not have symptomatic vision loss. [51] Patients with MS who have coexisting diabetes or a history of uveitis may be at an increased risk for development of FAME. [51] Dilated fundus examination, optical coherence tomography (OCT), and fluorescein angiography are the primary diagnostic tools in the evaluation of FAME. Treatment may consist of cessation of fingolimod therapy, observation, nonsteroidal anti-inflammatory agents, and/or corticosteroids. [51] Treatment typically starts with cessation of fingolimod treatment, which is often enough to alleviate manifestations of FAME. If development of FAME is suspected, referral of the patient to a general ophthalmologist, neuro-ophthalmologist, or retina specialist is appropriate.
Orbital disease
Alemtuzumab is a humanized monoclonal antibody that targets cells expressing CD52. As a disease-modifying therapy (DMT), this agent effectively decreases relapse rate and disability progression in patients with relapsing forms of multiple sclerosis MS (Table 2). However, secondary autoimmune disorders complicate therapy in nearly 50% of patients being treated with alemtuzumab, with Graves disease being the most common. [7, 53]
Affected patients may have mild to severe manifestations of Graves disease–related eye disease/orbitopathy characterized by proptosis, lid lag, lagophthalmos, corneal exposure, a restricted pattern of ocular misalignment, and optic nerve impingement. [7, 53] Thus, MS specialists treating patients with alemtuzumab should have a low threshold for referral of patients with suspected thyroid-related eye disease for consultation with an ophthalmologist.
Posterior visual pathway lesions
Lesions in the retrochiasmal and retrogeniculate visual pathways also affect patients with MS and other central nervous system (CNS) inflammatory disorders, albeit not with the same reported frequency as optic neuritis events. These lesions can be more difficult to diagnose because affected patients do not experience pain and may not be aware of deficits. If central vision is affected, patients with retrochiasmal or retrogeniculate lesions may describe missing parts of words of sentences with a binocular view, which is a hint that the visual deficit affects both eyes.
Vision loss is characterized by homonymous visual field deficits, which may or may not completely resolve over time. Homonymous defects can impair a patient’s ability to drive safely, particularly when he or she is unaware of the deficit. Furthermore, homonymous field deficits represent a potential red flag among patients with MS who use natalizumab, since they are at risk for development of progressive multifocal leukoencephalopathy (PML), a CNS infection caused by John Cunningham (JC) virus. This condition occurs more frequently in patients with MS who have previously used immunosuppressant drugs, who have serum positivity for JC virus, and who have a longer duration of immunosuppressant therapy. Natalizumab-associated PML is typically heralded by cognitive, motor, and language deficits; but vision loss and homonymous visual field deficits have been reported as presenting features. [52]
Progressive multifocal leukoencephalopathy has also been reported in the context of dimethyl fumarate use. [54] Therefore, new onset of homonymous visual field loss in patients with MS who have a history of treatment with natalizumab (or immunosuppression due to combination therapies or newer disease-modifying agents) should prompt consideration of PML, in part because of the high rates of morbidity and mortality associated with this diagnosis.
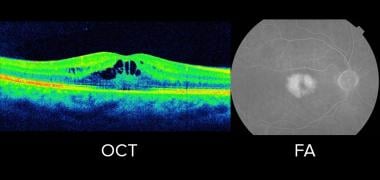 Optical coherence tomography (OCT) and fluorescein angiography (FA) showing cystoid macular edema in fingolimod-associated macular edema (FAME).
Optical coherence tomography (OCT) and fluorescein angiography (FA) showing cystoid macular edema in fingolimod-associated macular edema (FAME).
Efferent Visual Manifestations of Multiple Sclerosis
Patients with multiple sclerosis (MS) and CNS inflammatory disorders may present with ocular motility abnormalities on examination, although they may not report related symptoms.
Involvement of the brainstem and/or cerebellum can also cause damage to neural integrators in the CNS (medial vestibular nuclei, nucleus prepositus hypoglossus, interstitial nuclei of Cajal, and superior vestibular nuclei), [55] which help stabilize images on the retina during eye and head movements. This can lead to nystagmus, which is defined as repetitive to-and-fro involuntary eye movements initiated by slow drifts of the eye. [52, 56] Patients with nystagmus may note a perception that their world is moving, termed oscillopsia, and report blurred vision, imbalance, dizziness, and spatial disorientation. [52, 55, 56]
Patterns of nystagmus can be localized from an anatomic perspective and therefore help facilitate the diagnosis of MS vs other CNS inflammatory conditions. Nystagmus should not be confused with saccadic abnormalities that impair steady fixation.{Furthermore, patients may report focusing difficulties when watching objects in motion or when they themselves are in motion. These particular symptoms may arise from smooth pursuit abnormalities and impaired suppression of the vestibulo-ocular reflex. [55]
Efferent visual pathway lesions may be challenging to identify—and even more difficult to treat. To alleviate symptoms of diplopia, Fresnel prisms can be used temporarily to allow correction of ocular misalignment in primary position during the time required for recovery. In patients with persistent large-angle ocular deviations, ground-in prisms or, in rare cases, strabismus surgery may be feasible treatment options.
Diplopia
Ocular motor nerve and nuclear palsies
Inflammatory lesions in the brainstem may cause acquired ocular misalignment as a result of damage to the nuclei or fascicles of 1 of the 3 ocular motor nerves. Therefore, MS and other CNS inflammatory disorders should be considered in any young individual presenting with painless acute or subacute onset of diplopia.
Sixth cranial nerve: Nuclear and fascicular lesions
The paired abducens nuclei reside in the dorsal pons, separated from the floor of the fourth ventricle by the genu of the facial nerve (facial colliculus). [57] The abducens nucleus contains the neurons needed for horizontal gaze; interneurons traveling via the medial longitudinal fasciculus (MLF) connect the abducens nucleus with the contralateral oculomotor nucleus, coordinating abduction in one eye (governed by the lateral rectus) with adduction in the contralateral eye (medial rectus). [55, 57]
An abducens nuclear lesion causes horizontal gaze palsy ipsilateral to the side of the lesion, whereas a fascicular lesion causes ipsilateral abduction weakness with preserved adduction in the contralateral eye. Because of the proximity of the abducens nucleus to the facial nerve, patients may have a gaze palsy associated with a lower motor seventh nerve palsy, termed facial colliculus syndrome. [58] In one-and-a-half syndrome, an inflammatory lesion involving the abducens nucleus and the ipsilateral MLF causes an ipsilateral gaze palsy and ipsilateral internuclear ophthalmoplegia (INO). This causes either partial or complete loss of horizontal gaze function, except for the abduction in the eye opposite the one affected by INO, which may manifest features of abducting nystagmus.
Third cranial nerve: Nuclear and fascicular lesions
The oculomotor (third) nerve nuclear complex is located in the midbrain at the level of the superior colliculus. [57] Of the 4 paired subnuclei within this complex, the most medial innervates the superior rectus muscle. This is the only portion of the oculomotor nucleus that sends its axons to the opposite eye. [57] A nuclear third nerve lesion causes bilateral superior rectus weakness, bilateral ptosis, and, often, ipsilateral deficits reflective of third nerve fascicle dysfunction. In the case of a third nerve palsy, a fascicular lesion may cause partial or complete deficits in the following functions: ipsilateral elevation (superior rectus and inferior oblique muscles), depression (inferior rectus), and adduction of the eye (medial rectus); upper eyelid elevation; and pupillary constriction. [57] In rare cases, these lesions may be highly selective and may cause weakness of a single muscle, which can lead to diagnostic confusion.
The oculomotor nerve runs close to numerous midbrain structures; therefore, features of a third nerve palsy can be associated with other neurologic deficits, including contralateral hemiparesis (Weber syndrome), contralateral tremors (or chorea, athetosis) (Benedict syndrome), and contralateral cerebellar ataxia (Nothnagel syndrome). [57] Lesions of the dorsal midbrain in individuals with third nerve palsies may also manifest as paralysis of upgaze, light-near dissociation of the pupils, skew deviation, lid retraction, and convergence-retraction nystagmus. [57]
Fourth cranial nerve
The trochlear or fourth nucleus lies below the oculomotor nucleus, dorsal to the MLF, at the level of the inferior colliculus. [57] A lesion of the fourth nerve nucleus or proximal fascicle causes hyperdeviation (elevation) of the contralateral eye. The hyperdeviation of a fourth nerve palsy is greatest in contralateral gaze and is exacerbated by ipsilateral head tilt. A head tilt opposite the side of the higher or hyperdeviated eye tends to alleviate some symptoms. In patients with MS and other CNS inflammatory disorders, a demyelinating lesion may cause combined INO and contralateral fourth nerve palsy because of the anatomic proximity of the MLF and the fourth nerve nucleus and fascicle. [57] A trochlear nerve palsy may be difficult to distinguish from a skew deviation when the magnitude of hypertropia increases with ipsilateral head tilt.
In 2010, Wong proposed the “upright-supine test” to differentiate skew deviation from trochlear nerve palsy. During this clinical test, a near target is held one-third of a meter in front of the patient in both the upright and supine positions. The examiner looks for a vertical deviation that decreases by 50% or more from the upright to the supine position, suggesting skew deviation.{ref60 This should be ref 59 I think} Alternatively, if the upright-supine test result is negative and the vertical deviation decreases by less than 50% from the upright to the supine position, the vertical strabismus is likely caused by a fourth nerve palsy. [59]
Internuclear ophthalmoplegia
Internuclear ophthalmoplegia is a disconnection syndrome characterized by impaired horizontal gaze. Affected individuals have slowed or limited adduction in the eye ipsilateral to the lesion, with associated abducting nystagmus in the contralateral eye.{ref 52} [55] Unilateral or bilateral internuclear ophthalmoplegia is caused by damage to the MLFs. [55] The MLFs support the rapid neural transmission necessary for abduction of one eye and adduction of the fellow eye to be synchronous in horizontal gaze. Unlike other myelinated tracts in which slight impairment may produce no clinical deficits, the system for coordinated horizontal saccades is extremely sensitive to transmission speeds, making INO a common manifestation observed and reported by patients with MS. [52, 55, 60]
Patients with INO may deny symptoms, particularly with chronicity. Symptomatic patients may describe experiencing blurred vision, oscillopsia, and diplopia. Patients with INO may note worsening symptoms with fatigue or increased body temperature (Uhthoff phenomenon). [55] The reported variability and fatigability may be mistaken for the “pseudo-INO” of myasthenia gravis.
On clinical examination, the adduction deficit in a patient with INO may manifest as slowing during the horizontal duction (adduction lag with a full excursion of the eye) or as incomplete adduction producing an incomitant exotropia. The adduction lag in a patient with INO may be easily overlooked during smooth pursuit testing, and the clinician may need to provoke a quick horizontal saccade to unveil the disruption. Despite deficient adduction during horizontal saccades, medial rectus function is often intact in patients with INO; this feature can be demonstrated with testing of convergence, which is mediated by separate inputs to the medial rectus subnucleus, which are distinct from the inputs arriving via the MLF. [55, 57, 60]
The MLF also contains fibers that mediate vertical eye movements (pursuit, vestibular, and otolithic pathways), vertical gaze deficits may be observed. Patients with bilateral INO may have impaired vertical gaze-holding, resulting in primary-position or gaze-evoked vertical nystagmus. [55]
Variants of internuclear ophthalmoplegia
Wall-eyed internuclear ophthalmoplegia
Patients with unilateral INO do not typically have significant exotropia (outwardly deviating eye) in primary gaze because convergence tone is intact. [52, 55] In contrast, bilateral MLF lesions can cause exotropia in patients with wall-eyed bilateral INO (WEBINO) syndrome. [61] WEBINO syndrome differs from paralytic pontine exotropia of one-and-a-half syndrome, which is characterized by a unilateral horizontal gaze palsy and INO due to a pontine lesion involving the paramedian pontine reticular formation and MLF. [52, 55, 61]
One-and-a-half syndrome
One-and-a-half syndrome is a clinical disorder characterized by a conjugate horizontal gaze palsy in one direction with an associated INO. The syndrome usually results from a single unilateral lesion of the paramedian pontine reticular formation or the abducens nucleus on one side (causing the conjugate gaze palsy), with interruption of internuclear fibers of the ipsilateral MLF (causing failure of adduction of the ipsilateral eye). Consequently, the single preserved horizontal eye movement is abduction in the eye contralateral to the MLF lesion. [52]
Skew deviation
Skew deviation refers to a vertical ocular misalignment caused by supranuclear lesions disrupting inputs to the ocular motor nuclei. [54] Skew deviation may cause vertical diplopia or misalignment in patients with INO because the MLF contains utricular pathways that maintain vertical eye position in addition to interneurons from the abducens nucleus to the medial rectus subnucleus. [52, 61]
Ocular tilt reaction
Ocular tilt reaction consists of skew deviation, ocular torsion, head tilt, and deviation of the subjective visual vertical; all tilting is toward the lower or hypotropic eye. The side of the tilt is named for the lower eye. [62] A paroxysmal ocular tilt reaction is rare but has been described in patients with MS. [62]
Nystagmus
Nystagmus is common in patients with CNS inflammatory disorders, and it affects up to 30% of patients with MS. [52, 55] Common mechanisms that contribute to the development of nystagmus include impaired fixation, vestibular imbalance, and abnormal gaze-holding. [52, 55] Recognizing patterns of nystagmus can be useful in localizing lesions. Although much is known about the anatomy, physiology, and pharmacology of the ocular motor system, therapeutic options for nystagmus remain somewhat limited.
Table 6. Patterns of Nystagmus in Multiple Sclerosis and CNS Inflammatory Conditions [52, 55, 56, 63] (Open Table in a new window)
Type of Nystagmus |
Clinical Features |
Anatomic Site |
Treatments |
Gaze- evoked nystagmus |
Slow drift of eyes away from target followed by corrective saccade (jerk) in direction of eccentric gaze |
Lesions in brainstem or cerebellum that impair neural integrator function |
|
Pendular nystagmus |
Horizontal, vertical, or mixed components in one or both eyes with back-and-forth slow phase without corrective saccade (jerk) |
Brainstem or cerebellar lesions that damage neural integrator function |
Gabapentin, memantine, clonazepam |
Downbeat nystagmus |
Tonic upward deviation of eyes followed by fast downward saccade; may increase with downgaze and lateral gaze |
Lesion of cervicomedullary junction or cerebellum that disrupts input from posterior semicircular canals |
Clonazepam, baclofen gabapentin, 3,4-diaminopyridine, 4-aminopyridine, base-down prisms |
Upbeat nystagmus |
Downward drift of eyes followed by upward saccade; usually increases on upgaze; vertical smooth pursuit is usually disrupted by nystagmus |
Pontomedullary or pontomesencephalic lesions of ventral tegmental tract that disrupt projections from anterior semicircular canals |
Baclofen, aminopyridine |
Periodic alternating nystagmus |
Spontaneous horizontal beating nystagmus, the direction of which changes periodically; periods of oscillation range from 1 sec to 4 min (typically 1-2 min) |
Vestibulocerebellar damage; lesions of inferior cerebellar vermis that affect velocity storage mechanisms and stability of vestibulo-ocular reflex |
Baclofen, memantine |
Seesaw nystagmus |
Pendular or jerk oscillation characterized by an intorsion and elevation of one eye and a corresponding extorsion and depression of the other; during the next half-cycle, the torsional and vertical movements reverse |
Unilateral mesodiencephalic lesions that affect the interstitial nucleus of Cajal and vestibular afferents from the vertical semicircular canals (jerk hemi-seesaw); the pendular form of seesaw nystagmus is associated with lesions affecting the optic chiasm |
Clonazepam, memantine |
Saccadic abnormalities
Patients with central nervous system (CNS) inflammatory conditions including multiple sclerosis (MS) may demonstrate abnormal saccadic intrusions, causing vision loss and oscillopsia. [52, 55] Stable visual fixation is maintained by pause-cell neurons in the brainstem; these are located in the pontine raphe between the 2 abducens nuclei. [56] Pause cells prevent unwanted saccadic pulses by inhibiting saccadic premotor burst neurons located in the paramedian pontine reticular formation (PPRF) and the midbrain. [55, 56]
One example of a saccadic intrusion is the square wave jerk, which is a quick movement of the eye away from and back to primary position, which occurs with an intersaccadic latency of 150 to 200 milliseconds. [52, 55] Larger saccadic interruptions with a shorter intersaccadic latency (up to 80 milliseconds) are termed macro square wave jerks. [52, 55] Opsoclonus consists of repetitive bursts of conjugate saccadic oscillations with horizontal, vertical, and torsional elements. [52, 55] During each burst of these high-frequency oscillations, the movement is continuous without an intersaccadic interval. [52, 55]
In ocular flutter, there is no intersaccadic latency, although the pattern consists of saccadic eye movement restricted to the horizontal plane. [52, 55] Both opsoclonus and ocular flutter have been described in patients with MS. Variable treatment benefits have been reported with corticosteroids, thiamine, propranolol (40-80 mg orally 3 times daily), nitrazepam (15-30 mg orally daily), and clonazepam (0.5-2 mg orally 3 times daily). [56]
Patients with MS and other CNS inflammatory conditions also may present with features of hypometic and hypermetric saccades. [52, 55, 56]
Vestibular ocular reflex abnormalities
Some patients with central nervous system (CNS) inflammatory conditions including multiple sclerosis (MS) report difficulties tracking objects while they are in motion or when they are observing a moving target. Normally, during visual tracking, head movements accompany eye movements, with smooth pursuit allowing suppression of the normal vestibular ocular reflex. With inflammatory lesions of the cerebellar floccus, vestibular ocular reflex cancellation may be ineffective, which may cause loss of fixation during head and eye movement.Consequently, affected individuals experience retinal slippage off the target of interest, and compensatory catch-up saccades are produced. [52, 55, 64]
The integrity of the vestibular ocular reflex can be tested by asking the patient to perform rapid head thrusts while attempting to fixate on a stationary target. [52, 55] If vestibular ocular reflex function is impaired, fixation will not be maintained, and a compensatory saccadic eye movement opposite the direction of the head movement will be detectable. [52, 55]
Patients with CNS inflammatory syndromes including MS also are predisposed to impaired suppression of the normal vestibular ocular reflex. Normal suppression of the vestibular ocular reflex is needed to combine smooth head and eye movements during visual tracking. [52, 55, 64] To determine whether this problem exists clinically, a patient can be asked to focus on the thumb of his or her outstretched hand while rotating in a rotating chair. If vestibular ocular reflex cancellation is impaired, a series of catch-up saccades will be observed while the head is in motion. [52, 55, 64] The eyes will be observed to drift off the target (thumb) opposite the direction of the patient’s head movement. [52, 55] Vestibular rehabilitation therapy may prove beneficial for some affected individuals. [64]
Conclusions
Neuro-ophthalmologic manifestations are common in patients with multiple sclerosis (MS) and other central nervous system (CNS) inflammatory conditions, and it is therefore important to recognize the clinical manifestations of afferent and efferent visual pathway dysfunction in these patients.
Eye care specialists can play an important role in the multidisciplinary team approach required to oversee the care of affected individuals. Moreover, specialists in addition to ophthalmologists should be familiar with the clinical features and potential implications of visual symptoms in patients with MS and other CNS inflammatory syndromes; knowledge thereof can help guide selection of appropriate therapies and optimize surveillance efforts aimed at detecting disease activity and progression in patients with these conditions.
Questions & Answers
Overview
What are the neuro-ophthalmic manifestations of multiple sclerosis (MS)?
What is included in patient education about visual disturbances in multiple sclerosis (MS)?
What is multiple sclerosis (MS)?
What are the diagnostic criteria for multiple sclerosis (MS)?
What are the disease-modifying therapies used in the treatment of multiple sclerosis (MS)?
What is nystagmus in multiple sclerosis (MS)?
What are ocular motility abnormalities in multiple sclerosis (MS)?
What is the role of multiple sclerosis (MS) in the development of diplopia?
What are the sixth cranial nerve nuclear and fascicular lesions in multiple sclerosis (MS)?
What are the third cranial nerve nuclear and fascicular lesions in multiple sclerosis (MS)?
What are the fourth cranial nerve nuclear and fascicular lesions in multiple sclerosis (MS)?
What is internuclear ophthalmoplegia (INO) in multiple sclerosis (MS)?
What are the variants of internuclear ophthalmoplegia (INO) in multiple sclerosis (MS)?
What are saccadic abnormalities in multiple sclerosis (MS)?
What are vestibular ocular reflex abnormalities in multiple sclerosis (MS)?
What is optic neuritis in multiple sclerosis (MS)?
Which clinical history findings are characteristic of optic neuritis in multiple sclerosis (MS)?
Which physical findings are characteristic of optic neuritis in multiple sclerosis (MS)?
How does pediatric optic neuritis in multiple sclerosis (MS) differ from the adult-onset syndrome?
How is optic neuritis in multiple sclerosis (MS) diagnosed?
What is the role of lab tests in the workup of optic neuritis in multiple sclerosis (MS)?
How is optic neuritis in multiple sclerosis (MS) treated?
What is the prognosis of optic neuritis in multiple sclerosis (MS)?
What is uveitis in multiple sclerosis (MS)?
What are posterior visual pathway lesions in multiple sclerosis (MS)?
-
Magnetic resonance imaging (MRI) scan of the head of a 35-year-old man relapsing-remitting multiple sclerosis. MRI reveals multiple lesions with high T2-signal intensity and 1 large white matter lesion. In some cases, these demyelinating lesions mimic brain tumors because of the associated edema and inflammation.
-
Normal-appearing right optic nerve and mild left optic disc edema.
-
The evolution of temporal optic disc pallor (arrow) after left optic neuritis.
-
Cranial MRI study showing typical white matter lesions (arrows).
-
Optical coherence tomography (OCT) and fluorescein angiography (FA) showing cystoid macular edema in fingolimod-associated macular edema (FAME).
Tables
- Table 1. The 2017 McDonald Criteria for Diagnosis of MS in Patients with an Attack at Onset (modified from Thompson et al 2017) [6]
- Table 2. Approved Disease-Modifying Therapies (DMTs) Used in the Treatment of Multiple Sclerosis [7, 8, 9, 10, 11, 12, 13]
- Table 3. NMOSD Diagnostic Criteria for Adult Patients [14]
- Table 4. Clinical, Serologic, And Radiologic Features Distinguishing MS, NMOSD, and MOGAD [6, 10, 14, 17, 18, 20, 24, 25]
- Table 5. Differential Diagnoses of Optic Neuritis (Modified from Table 2-1 in Costello F. Inflammatory optic neuropathies. Continuum (Minneap Minn). Aug 2014; 20 (4 Neuro-ophthalmology): 816-37.] [28]
- Table 6. Patterns of Nystagmus in Multiple Sclerosis and CNS Inflammatory Conditions [52, 55, 56, 63]
Number of Clinical Attacks |
Number of Lesions with Objective Clinical Evidence |
Additional Data Required for MS Diagnosis |
≥ 2 |
≥ 2 |
None |
≥ 2 |
1 (plus clear evidence of a previous attack involving a lesion in a distinct neuroanatomic location) |
None |
≥ 2 |
1 |
DIS demonstrated by an additional clinical attack implicating a different CNS location or by MRI a |
1 |
>2 |
DIT demonstrated by an additional clinical attack or by MRI b OR demonstration of CSF OCBs |
1 |
1 |
DIS demonstrated by an additional clinical attack implicating a different CNS site or by MRIa AND DIT shown by an additional clinical attack or by MRIb OR demonstration CSF OCBs |
Abbreviations: CSF = cerebrospinal fluid; CNS = central nervous system; DIS = dissemination in space; DIT = dissemination in time; MRI = magnetic resonance imaging; OCBs = oligoclonal bands
Magnetic Resonance Imaging (MRI) Criteria: aMRI DIS can be indicated by 1 or more T2- hyperintense lesions that are characteristic of MS in 2 or more of the 4 areas of the CNS: periventricular, cortical or juxtacortical, infratentorial, and spinal cord. bMRI DIT can be indicated by the simultaneous presence of gadolinium-enhancing and nonenhancing lesions at any time or by a new T2-hyperintense or gadolinium-enhancing lesion on follow-up MRI, with reference to a baseline scan, regardless of the timing of the baseline scan. |
||
Drug |
Route |
MS Subtype |
Dosing Frequency |
Most Common Adverse Effects |
Interferon β-1a (Avonex) |
IM |
Relapsing forms of MS, CIS |
Once weekly |
Flulike symptoms Liver enzyme changes BMS Thyroid dysfunction |
Interferon β-1a (Rebif) |
SC |
Relapsing forms of MS, CIS |
22 μg or 44 μg 3 times weekly |
Flulike symptoms Liver enzyme changes BMS Thyroid dysfunction |
Interferon β-1b (Betaseron) (Extavia) |
SC |
Relapsing forms of MS, CIS |
3 times weekly |
Flulike symptoms Liver enzyme changes BMS Thyroid dysfunction |
Glatiramer acetate (Copaxone) Glatopa (glatiramer acetate–generic equivalent of Copaxone 20mg and 40 mg doses) |
SC |
Relapsing forms of MS, CIS |
Once daily |
Skin irritation Skin lipoatrophy Panic attack–like events (often self limited) |
| Peginterferon beta 1a (Plegridy) | SC or IM | Relapsing forms of MS, CIS | Every 2 weeks | Flulike symptoms Liver enzyme changes Depression/suicide Injection site reactions Low peripheral blood counts CHF Seizures Thrombotic microangiopathy Autoimmune disorders |
Natalizumab (Tysabri) |
IV |
Relapsing forms of MS, CIS |
Once monthly |
Nausea Infection Liver dysfunction Binding antibodies PMLa |
Fingolimod (Gilenya) |
PO |
Relapsing forms of MS, CIS |
Once daily |
Macular edema Bradyarrhythmia QT interval prolongation HTN Severe varicella-associated complications in nonimmune patients Increased risk for HZ in all patients Mild infections PML |
Dimethyl fumarate (Tecfidera) |
PO |
Relapsing forms of MS, CIS |
Twice daily |
Flushing GI distress Lymphopenia PML (rare) |
| Monomethyl fumarate (Bafiertam) | PO | Relapsing forms of MS, CIS | Twice daily | Flushing GI illness HZ, opportunistic infections Lymphopenia PML (rare) |
Teriflunomide (Aubagio) |
PO |
Relapsing forms of MS, CIS |
Once daily |
Nausea Headaches Alopecia Liver dysfunction Presumed teratogenicity |
Alemtuzumab (Lemtrada) |
IV | Relapsing forms of MS |
Minimum of 2 cycles (baseline and Year 1) |
Infusion reactions Mild to moderate infections Thyroid dysfunction ITP Anti–GBM disease Stroke Alveolar hemorrhage Autoimmune hemolytic anemia Hemophagocytic lymphohistiocytosis |
Ocrelizumab (Ocrevus) |
IV |
Relapsing forms of MS, CIS |
Every 6 months |
Hypogammaglobulinemia Infections (skin, urinary, sinus) Infusion reactions Rare neutropenia and lymphopenia Uncertain if true link to breast cancer |
Ofatumumab (Kesimpta) |
SC |
Relapsing forms of MS, CIS |
Once monthly |
Injection site reactions URIs UTIs Headache Reduced IgM |
Cladribine (Mavenclad) |
PO | Relapsing forms of MS |
Yearly (for 2 years) |
Headaches Lymphopenia HZ infections Nausea Abdominal pain Bronchitis Diarrhea Increased risk for routine infections |
Siponimod (Mayzent) |
PO |
Relapsing forms of MS, CIS |
Once daily |
Lymphopenia Increased LFTs Rare bradycardia Macular edema HTN Varicella zoster infection |
Ozanimod (Zeposia) |
PO |
Relapsing forms of MS, CIS |
Once daily |
URIs Hepatic transaminase elevations Orthostatic hypotension UTIs Back pain HTN |
| Ponesimod (Ponvory) | PO | Relapsing forms of MS, CIS | Once daily | Infections Bradyarrhythmia Liver enzyme abnormalities HTN |
| Diroximel fumarate (Vumerity) | PO | Relapsing forms of MS, CIS | Twice daily | Flushing GI illness HZ, opportunistic infections Lymphopenia PML (rare) |
Abbreviations: CIS = clinically isolated syndrome; MS = multiple sclerosis; GI = gastrointestinal; HZ = herpes zoster; CHF = congestive heart failure; HTN = hypertension; URI = upper respiratory tract infection; UTI = urinary tract infection; BMS = bone marrow suppression; ITP = Idiopathic thrombocytopenic purpura; Anti-GBM disease = anti–glomerular basement membrane disease; LFT = liver function test; PML = progressive multifocal leukoencephalopathy. aRisk of PML risk increases with a history of immunosuppression, JC virus positivity, and use of the drug for more than 2 years. |
||||
|
NMOSD with + AQP4-IgG
|
NMOSD with AQP4-IgG Negative/Unknown Status
At least 2 core clinical characteristics resulting from 1 or more clinical attacks, meeting the following requirements:
|
Core Clinical Characteristics
|
a Additional MRI Requirements
|
| Abbreviations: AQP4-IgG = aquaporin 4–IgG ; MRI = magnetic resonance imaging; NMOSD = neuromyelitis optica spectrum disorders |
Feature |
MS |
NMOSD |
MOGAD |
Age |
Variable (median age at onset in 3rd decade) |
Variable (median age at onset > age at onset of MS and in 4th decade) |
Variable |
Female: Male |
2-3:1 |
9:1 |
1.5:1 |
Epidemiology |
White persons more often affected |
Nonwhite persons including Black persons and persons of Asian and Afro-Caribbean descent more predisposed |
Unknown |
Serologic Marker |
None |
AQP4-IgG |
MOG-IgG |
Brain MRI |
Ovoid periventricular, juxtacortical, cortical, infratentorial peripheral, ring-/open ring-enhancing T2, FLAIR, Gd lesions |
Can be normal or show nonspecific lesions; classic lesions may also be found in the area postrema, region of the 3rd/4th ventricle, splenium of the corpus callosum (20% of patients may have MS-appearing lesions) |
Lesions that are ADEM-like, involving the deep gray matter, appearing diffuse/ confluent in the brainstem, affecting cerebellar peduncles |
Optic Nerve MRI |
Often unilateral, involving short segments (< 50%) of affected nerve length |
Often bilateral with enhancement of >50% of optic nerve, particularly involving posterior regions of the anterior visual pathway including chiasm |
Often bilateral with enhancement of >50% of optic nerve, particularly intraorbital optic nerve with perineural involvement |
Spine MRI |
Multiple lesions may be seen including short lesions; periphery of cord (dorsal/lateral column); ring or variable enhancement |
Single lesion (LETM) and short areas of segmental involvement may be seen with variable enhancement |
Multiple lesions may be seen (75% LETM; 25% short); the conus may be involved and lesional enhancement is variable |
Acute Treatment |
High-dose corticosteroids (rarely plasma exchange) |
High-dose corticosteroids ± plasma exchange |
High-dose corticosteroids ± plasma exchange/IVIG (in children) |
Long-term Management |
DMTs |
Azathioprine, mycophenolate mofetil, rituximab; newer targeted agents include eculizumab, satralizumab, and inebilizumab |
Options for recurrent disease include azathioprine, mycophenolate mofetil, rituximab |
|
|||
Diagnosis |
Clinical Features |
Investigations to Consider |
NA-AION |
Painless, altitudinal visual field defect common; vision loss on awakening; vascular risk factors include phosphodiesterase type 5 inhibitor use, nocturnal antihypertensive use, sleep apnea; physiologic disc at risk, patients with NA-ION have optic disc edema acutely |
Sleep study, 24h BP monitoring, investigations for HTN and diabetes |
Compressive optic neuropathy (pituitary lesions, meningiomas, aneurysm) |
Painless, progressive vision loss; color loss disproportionate to visual acuity deficit; non-glaucomatous optic disc cupping; temporal visual field cut; bilateral visual field involvement |
Cranial and orbital MRI/MRA or CT/CTA |
Infectious optic neuropathies (eg, tuberculosis, syphilis, Lyme disease) |
Associated uveitis, papillitis, or retrobulbar optic neuropathy; macular star; infectious symptomatology |
Serum/CSF culture/sensitivity; specific serologic testing for syphilis, Lyme, Bartonella henselae, HIV, toxoplasmosis, HBV and HCV; EBV; histoplasmosis; TB testing; chest imaging; ESR, CRP |
Inflammatory/demyelinating optic neuropathies not associated with MS or an underlying systemic disorder: NMOSD, CRION, ADEM, and MOGAD |
Poor recovery, unilateral or bilateral optic neuritis, associated transverse myelitis, recurrent symptoms |
Brain MRI, cervical spine MRI, serum anti-AQP4 IgG antibody testing, serum anti-MOG IgG antibody testing |
Genetic optic neuropathies (LHON, autosomal-dominant optic neuropathy) |
Bilateral vision loss, painless, poor recovery, family history |
Genetics referral with specific mutation testing |
Toxic/nutritional (tobacco-alcohol amblyopia and Cuban and Tanzanian epidemic optic neuropathies) |
Bilateral optic nerve involvement, history of drug use (ethambutol, selenium, amiodarone), restricted nutritional intake, glue sniffing, methanol ingestion |
Vitamin B12 levels, toxicology screen |
Sarcoid optic neuropathy |
Steroid responsive, poor recovery, systemic symptoms and signs |
Chest imaging, serum ACE, gallium scan, tissue diagnosis, bronchoalveolar lavage, soluble IL-2 receptor |
Connective tissue/vasculitic optic neuropathy (lupus, Wegener granulomatosis, Sjögren syndrome, Behçet disease) |
Steroid responsive, associated systemic symptoms and signs |
Serum ESR, Sjögren-specific antibodies, CRP, ANCA, ENA panel, ANA |
Orbital inflammation/optic perineuritis |
Orbital signs (proptosis) |
MRI or CT orbital imaging, blood work including TSH ANCA, CRP, ESR, ACE |
Uveitis/posterior scleritis |
Severe pain, floaters, vitreous reaction |
Fluorescein angiography, B-scan ultrasonography of orbits |
Autoimmune optic neuropathy (similar to CRION) |
Steroid responsive |
Skin biopsy for immunoglobulin deposition |
Big blind spot syndromes |
Blind spot on visual field testing, painless photopsias, bilateral ocular involvement |
Full-field/multifocal ERG, fluorescein angiography |
Abbreviations: ACE = angiotensin-converting enzyme; ADEM = acute disseminated encephalomyelitis; anti-DS DNA = anti-double-stranded DNA; anti-MOG = anti–myelin oligodendrocyte glycoprotein; ANA = antinuclear antibody; ANCA = antineutrophilic cytoplasmic antibody; AON = autoimmune optic neuropathy; AQP4 = aquaporin 4; CRION = chronic relapsing inflammatory optic neuropathy; CRP = C-reactive protein; CSF = cerebrospinal fluid; CT = computed tomography; CTA = computed tomographic angiography; ENA = extractable nuclear antigen; ERG = electroretinogram; ESR = erythrocyte sedimentation rate; FTA-ABS = fluorescent treponemal antibody absorption; LHON = Leber hereditary optic neuropathy; MOG = myelin oligodendrocyte glycoprotein; MOGAD = myelin oligodendrocyte glycoprotein IgG-associated disease; MRA = magnetic resonance angiography; MRI = magnetic resonance imaging; MS = multiple sclerosis; NA-ION = nonarteritic anterior ischemic optic neuropathy; NMO-IgG = neuromyelitis optica IgG; NMSOD = neuromyelitis optica spectrum disorder; PCR = polymerase chain reaction; TSH = thyroid-stimulating hormone; VDRL = Venereal Disease Research Laboratory; ; HBV = hepatitis B virus; HCV = hepatitis C virus; EBV = Epstein-Barr virus; ESR = serum sedimentation rate; CRP = C-reactive protein |
||
Type of Nystagmus |
Clinical Features |
Anatomic Site |
Treatments |
Gaze- evoked nystagmus |
Slow drift of eyes away from target followed by corrective saccade (jerk) in direction of eccentric gaze |
Lesions in brainstem or cerebellum that impair neural integrator function |
|
Pendular nystagmus |
Horizontal, vertical, or mixed components in one or both eyes with back-and-forth slow phase without corrective saccade (jerk) |
Brainstem or cerebellar lesions that damage neural integrator function |
Gabapentin, memantine, clonazepam |
Downbeat nystagmus |
Tonic upward deviation of eyes followed by fast downward saccade; may increase with downgaze and lateral gaze |
Lesion of cervicomedullary junction or cerebellum that disrupts input from posterior semicircular canals |
Clonazepam, baclofen gabapentin, 3,4-diaminopyridine, 4-aminopyridine, base-down prisms |
Upbeat nystagmus |
Downward drift of eyes followed by upward saccade; usually increases on upgaze; vertical smooth pursuit is usually disrupted by nystagmus |
Pontomedullary or pontomesencephalic lesions of ventral tegmental tract that disrupt projections from anterior semicircular canals |
Baclofen, aminopyridine |
Periodic alternating nystagmus |
Spontaneous horizontal beating nystagmus, the direction of which changes periodically; periods of oscillation range from 1 sec to 4 min (typically 1-2 min) |
Vestibulocerebellar damage; lesions of inferior cerebellar vermis that affect velocity storage mechanisms and stability of vestibulo-ocular reflex |
Baclofen, memantine |
Seesaw nystagmus |
Pendular or jerk oscillation characterized by an intorsion and elevation of one eye and a corresponding extorsion and depression of the other; during the next half-cycle, the torsional and vertical movements reverse |
Unilateral mesodiencephalic lesions that affect the interstitial nucleus of Cajal and vestibular afferents from the vertical semicircular canals (jerk hemi-seesaw); the pendular form of seesaw nystagmus is associated with lesions affecting the optic chiasm |
Clonazepam, memantine |








