Background
Bovine spongiform encephalopathy (BSE), also known as mad cow disease, and variant Creutzfeldt-Jakob disease (CJD) are related disorders. [1] They belong to the family of diseases known as the transmissible spongiform encephalopathies (TSEs). TSEs are caused by a transmissible proteinaceous particle, which is yet to be fully characterized. Other TSEs include scrapie (a disease of sheep), feline spongiform encephalopathy, transmissible mink encephalopathy, and chronic wasting disease of deer and elk. Human forms include classic CJD, variant CJD, kuru, Gerstmann-Sträussler-Scheinker disease, familial fatal insomnia, and sporadic fatal insomnia. [2, 3, 4, 5, 6, 7, 8]
Human TSEs share the following characteristics:
-
A prolonged incubation period of several years
-
A progressive debilitating neurologic syndrome that is invariably fatal
-
Pathological changes that are confined to the CNS and consist of the following 3 classic features: spongiosis, gliosis, and neuronal loss
-
A transmissible agent that does not elicit any specific immunologic response in the host and is unusually resistant to conventional inactivation procedures
Bovine spongiform encephalopathy
On December 23, 2003, targeted surveillance identified a “downer” dairy cow (ie, nonambulatory and disabled) that tested positive for BSE. This was confirmed by the BSE International Reference Laboratory in Weybridge, England, on December 25. On December 9, 2003, the downer dairy cow had been slaughtered in the state of Washington. Because the animal's condition was attributed to complications from calving, the meat was considered safe for human consumption by the US Department of Agriculture (USDA). Tissues such as brain, spinal cord, and small intestine, which may have a higher likelihood of containing the pathogenic agent of BSE, were removed during slaughter and sent for rendering (often to be used as nonruminant animal feed).
Not surprisingly, international reaction was swift. Within a week, 53 countries had imposed a ban on imports of US beef and beef products. On December 30, the USDA announced new rules banning all downer cattle from the chain of human food production and other measures. Subsequently, the infected cow was discovered to have originated in Alberta, Canada, and was imported into the United States in September 2001. [9]
On January 26, 2004, the US Food and Drug Administration (FDA) announced new rules to further strengthen existing protection against BSE, including banning a wide range of bovine material from human food (United States Department of Health and Human Services, Expanded "Mad Cow" Safeguards Announced To Strengthen Existing Firewalls Against BSE Transmission). On February 9, 2004, the USDA completed its investigations.
Although this was the first case of BSE in the United States, more than 190,000 confirmed clinical cases have been reported worldwide since 1986, and approximately 184,000 cases were from the United Kingdom alone (see the images below). Additional information is available at OIE, Bovine Spongiform Encephalopathy (BSE). Based on mathematical modeling of the BSE epidemic, estimates suggest that 1-3 million cattle may have been infected with the BSE agent in the United Kingdom. [10, 11] Most of these infected animals were slaughtered for human consumption before any clinical signs of BSE were noted.
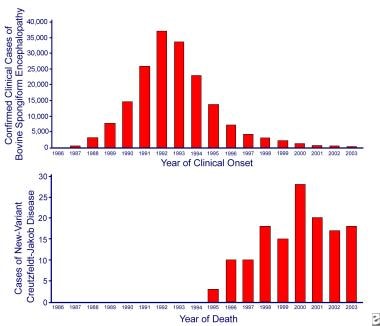 Incidence of bovine spongiform encephalopathy (BSE) and variant Creutzfeldt-Jakob disease (CJD) in Great Britain. The BSE epidemic peaked in 1992, 4 years after the introduction of the ban on ruminant feed. The associated human disease, variant CJD, was not defined until 1996, 7 years after a ban was introduced in Britain on the use of specified offal from cattle in human food.
Incidence of bovine spongiform encephalopathy (BSE) and variant Creutzfeldt-Jakob disease (CJD) in Great Britain. The BSE epidemic peaked in 1992, 4 years after the introduction of the ban on ruminant feed. The associated human disease, variant CJD, was not defined until 1996, 7 years after a ban was introduced in Britain on the use of specified offal from cattle in human food.
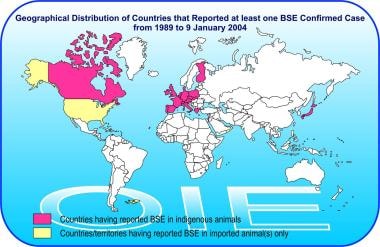 Geographic distribution of bovine spongiform encephalopathy (BSE) by country as of January 9, 2004. From http://www.oie.int/eng/info/en_esb.htm.
Geographic distribution of bovine spongiform encephalopathy (BSE) by country as of January 9, 2004. From http://www.oie.int/eng/info/en_esb.htm.
Other countries where BSE has been confirmed in native-born cattle include Austria, Belgium, Canada, Czech Republic, Denmark, Finland, France, Germany, Greece, Ireland, Israel, Italy, Japan, Luxembourg, Liechtenstein, the Netherlands, Poland, Portugal, Slovakia, Slovenia, Switzerland, and Spain. Additional cases of BSE have also been confirmed in North America: 9 in Canada (one cow was imported from the United Kingdom) and 3 in the United States (2 were imported from Canada). The third case of BSE reported from the United States was in a downer cow on a farm in Alabama, and the herd of origin could not be identified in spite of a thorough investigation. [12]
A fourth case in the United States (the first since 2006) was detected as part of a screening program in California and did not enter the food chain. [13]
Further cases of BSE were reported in imported cattle in the Falkland Islands (imported from the United Kingdom) and Oman (imported from the United Kingdom). No documented cases have been reported from Africa, Australia, New Zealand, or South America.
Approximately 5 million head of cattle were slaughtered in the United Kingdom to halt the epidemic. Since 1992, the number of cases has decreased an average of 40% per year, although new cases continue to be reported. However, the preemptive slaughter crippled the British livestock industry and affected the tallow, gelatin, and pharmaceutical industries. [14]
The incubation period for BSE ranges from 2-8 years. Most cases in the United Kingdom have occurred in dairy cows aged 3-6 years. The clinical features include the following:
-
Changes in temperament such as nervousness or apprehensiveness
-
Aggression toward other cattle or humans
-
Kicking when being milked
-
Reluctance to cross concrete, turn corners, and enter yards or doorways
-
Head shyness with head held low
-
Abnormal posture
-
High-stepping gait, particularly of hind legs
-
Incoordination
-
Difficulty in rising or walking
-
Skin tremors
-
Decreased milk production
-
Weight loss despite good appetite
No treatment is available for BSE; the disease is relentlessly progressive until the animal dies or is destroyed. This usually occurs in 2 weeks to 6 months.
No test can detect the disease in a live animal, although in an epidemic setting, clinical features are sufficiently distinctive to provide a clinical diagnosis. Currently, 3 laboratory methods are used to confirm the diagnosis of BSE, including the following:
-
Immunohistochemical labeling of the disease-associated (abnormal) prion protein (PrPSc)
-
Detection of scrapie-associated fibrils (SAF) by electron microscopy
Hypotheses of origin
Different hypotheses are proposed regarding the origins of BSE. The most compelling hypothesis is that BSE originated from scrapie, an endemic spongiform encephalopathy of sheep and goats that has been endemic in Europe since the mid-18th century. [17] Scrapie has since spread to most sheep-breeding countries and is widespread in the United Kingdom, where, until 1988, the rendered carcasses of livestock (including sheep) were fed to ruminants and other animals as a protein-rich nutritional supplement. The epidemiologic data appear to implicate feed containing TSE-contaminated meat and bone meal, which was used as a protein source. The causative agent is suspected to be from either scrapie-affected sheep or cattle with previously unidentified TSE. [18, 19, 20, 21]
Changes in the rendering process that took place in the early 1980s, particularly the removal of a solvent extraction process that included a steam heat treatment, probably allowed the etiologic agent to survive, contaminate the protein supplement, and infect cattle. Recycling of infected bovine carcasses within the cattle population (turning the herbivorous cows into "animal cannibals") amplified the levels of the pathogen, which had become adapted to cattle, in the feeds and eventually caused a full-scale epizootic. [22] Similarly, the spread of spongiform encephalopathies in farmed mink and captive and zoo animals may have resulted from prion-contaminated feed. [23, 24]
An alternative hypothesis was proposed in the controversial final BSE inquiry report in the United Kingdom that was released October 24, 2000, suggesting that a pathogenic mutation occurred in cattle in the 1970s, with BSE occurring as a consequence of recycling of infected cattle. The report asserts that BSE cases identified from 1986-1988 were not index cases, nor were they the result of the transmission of scrapie.
Recognition of the source of infection led to several countermeasures to break the cycle of cattle reinfection, restrict the geographic spread, and eliminate the potential source of new infection. The most important step was banning ruminant feed in 1988, extending it to include the feeding of specified bovine offal. By 1992, this ban started to bring the epidemic under control. See the image below.
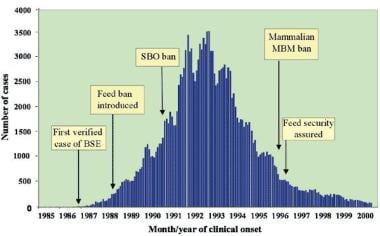 Time course of epidemic bovine spongiform encephalopathy (BSE) in the United Kingdom, 1986-2000, with dates of major precautionary interventions. SBO stands for specified bovine offal (ie, brain, spinal cord, thymus, spleen, and intestines from cattle aged >6 mo). MBM stands for meat and bone meal (protein residue produced by rendering). From Brown P, Will RG, Bradley R, et al. "Bovine spongiform encephalopathy and variant Creutzfeldt-Jakob disease: background, evolution and current concerns". Emerging Infectious Diseases, 2001;7: 6-16.
Time course of epidemic bovine spongiform encephalopathy (BSE) in the United Kingdom, 1986-2000, with dates of major precautionary interventions. SBO stands for specified bovine offal (ie, brain, spinal cord, thymus, spleen, and intestines from cattle aged >6 mo). MBM stands for meat and bone meal (protein residue produced by rendering). From Brown P, Will RG, Bradley R, et al. "Bovine spongiform encephalopathy and variant Creutzfeldt-Jakob disease: background, evolution and current concerns". Emerging Infectious Diseases, 2001;7: 6-16.
Furthermore, specified risk material, which comprises brain, spinal cord, eyes, tonsils, thymus, spleen, and intestine, is removed from all foodstuffs at slaughter. [25] In addition, cattle aged 30 months or older must not be used for consumption unless they test negative for BSE, which is known as the “over-thirty-month” rule. [26]
Variant CJD
Within weeks of identification of the first case of BSE, concern was expressed about human risk. [27, 28, 29] A national TSE surveillance was instituted in Britain in 1990 despite lack of evidence of human acquisition of scrapie based on the then-speculative grounds that exposure of millions of Britons to an apparently new bovine TSE might unmask low-frequency transmission in humans. Unfortunately, this fear proved to be well founded. The first cases of variant CJD (initially called new variant CJD) were reported in 1995. [30, 31]
By 1996, 10 patients, who had distinctive clinical and neuropathologic characteristics, had been reported to the National CJD Surveillance Unit with atypical CJD-like features. Clinically, they were younger than 40 years, with an average age of younger than 30 years; had behavioral symptoms, ataxia, and sensory changes early on; and progressed more slowly than in classic CJD. None had periodic complexes on EEG. Neuropathologic findings resembled those of kuru, with extensive florid plaques in which an amyloid core is surrounded by petals of spongiform change. The report concluded that this hitherto unrecognized variant of CJD was probably due to exposure to BSE. [32] So far, 217 cases of variant CJD have been described, most from the United Kingdom. [6]
The risk factors for the development of variant CJD include younger age (as compared with classic CJD), residence in the United Kingdom, and methionine homozygosity at codon 129 of the prion protein gene (PRNP). [33, 34] The encoding alternatives, methionine (Met) and valine (Val), are distributed in white populations in the approximate proportions of 50% Met/Val, 40% Met/Met, and 10% Val/Val. All patients with variant CJD who have been tested have been homozygous for methionine. [35] A reduced frequency of HLA class II type DQ7 has been described in patients with variant CJD but not in those with classic CJD; this may have important implications for understanding host susceptibility to infection by BSE prions. [36] Past surgery, previous blood transfusion, and occupation have not been shown to be associated with increased risk, although 2 cases have been reported in patients who received blood transfusions from donors who then went on to develop variant CJD.
Experiments in transgenic mice have shown that a significant species barrier exists that restricts the transmission of BSE to humans; however, the barrier is significantly reduced for human-to-human transmission, with increasing transmission efficiency from Met/Met to Met/Val to Val/Val genotypes. [37]
Pathophysiology and Etiology
Convincing evidence indicates that variant Creutzfeldt-Jakob disease (CJD) is a new disease. Despite its name, variant CJD appears to be a human variant of BSE derived from a cow-to-human species switch, rather than an actual variant of human sporadic CJD. [38] Epidemiologic, biological, and biochemical data favor the hypothesis that variant CJD is a BSE zoonosis, probably arising from a double-species switch from sheep scrapie to BSE and then from bovine spongiform encephalopathy (BSE) to human variant CJD. [38, 39, 40]
Although the BSE epizootic has apparently led to other newly host-switched transmissible spongiform encephalopathies (TSEs) in domestic and large cats, sheep who have been fed the BSE agent experimentally have acquired a scrapie-like disease. [41] Such occurrences widen the scope of possible TSE species switches and back-switches and suggest that the BSE agent may be an uncharacteristically promiscuous prion. [38]
BSE and variant CJD are similar on the basis of patterns of infectable mouse strains, incubation time, survival time, lesion distribution in the mouse brain, PrPSc gel banding patterns, and neuropathology, which are readily distinguishable from other TSEs, such as scrapie and sporadic CJD. [42, 43, 44, 45, 46] Pathological investigation shows characteristic spongiform change and gliosis in the brain. These changes are most predominant in basal ganglia and cerebellum.
Because no occupational exposure of patients with variant CJD to cattle on farms or in abattoirs has been identified, spread is likely to occur through consumption of BSE-contaminated meat products, yet there was a case reported in 2020 of suspected occupational infection in a laboratory worker. [47] Whether PrPSc (prion protein, scrapie isoform) can be demonstrated in skeletal muscles remains controversial. [48, 49, 50, 51] However, a high-sensitivity Western blotting technique identified muscle PrPSc in 8 of the 17 patients studied, although in much lower concentrations than in the cerebral cortex, suggesting a potential role for skeletal muscle in the transmission of variant CJD. [52] Despite this evidence, the infection probably resulted from beef products contaminated by nervous tissue, because neural tissues have a much higher concentration of PrPSc than other peripheral tissues.
The amount of infectious agent ingested and host susceptibility, as determined by the human genotype at PRNP codon 129, appear to play important roles in the development of variant CJD. However, how oral consumption of BSE-contaminated beef leads to infection of the CNS is unknown. In the early preclinical stages of the disease, PrPSc can be detected in lymphoid tissues, suggesting a possible route of transmission from the gut. Prions probably cross the mucosa via transmembranous tunneling of the membranous epithelial cells (M-cells) and come in contact with the mucosa-associated lymphoid system, including Peyer patches, where accumulation is found first. [53]
A functional immune system is required for prion replication and transport outside of the CNS. [54] Mechanisms of further prion transport to other compartments of the lymphoreticular system (LRS) are unclear. Prions accumulate in cells of the LRS, most prominently in the follicular dendritic cells and in sympathetic nerve endings in the LRS. Then, prions reach the CNS via splanchnic nerves at the level of the thoracic spinal cord and via parasympathetic fibers connecting with the brain. [55, 56] The other possible route is blood, which was suggested by experiments showing BSE transmission from sheep to sheep by blood transfusion. [57]
Variant CJD is known to affect the brain, lymphoreticular system, pituitary and adrenal glands, and gastrointestinal tract. One case report identified protease-resistant prion protein in the dura mater, liver, pancreas, kidney, ovary, uterus, and skin of a patient with variant CJD, indicating that organ involvement may actually be even more widespread. [58]
Variant CJD in the blood supply
Concern is widespread that the blood supply might be contaminated with the variant CJD agent. This possibility is supported by evidence that BSE in sheep can be transmitted by blood transfusion. [57] This concern progressed to fear with a report of a patient who died of variant CJD 6.5 years after receiving a transfusion of red blood cells donated by an individual who subsequently developed variant CJD. [59] The authors do not present direct evidence that the disease was transmitted by blood transfusion, but the chance that this case is not transfusion related is very small.
One more case of blood transfusion–related variant CJD has been reported. [60] This case is unique and very important in terms of its implications. This patient died from a non-neurologic disorder 5 years after receiving a blood transfusion from a donor who subsequently developed variant CJD, and he had no symptoms suggestive of variant CJD at the time of his death. Protease-resistant prion protein (PrPres) was detected in the spleen and a cervical lymph node but not in the brain. He was heterozygote at codon 129 of PRNP, suggesting that susceptibility to variant CJD infection is not confined to methionine homozygous PRNP genotype.
In the late 1990s, the United Kingdom implemented strategies for risk reduction. These included importation of plasma from the United States for preparation of plasma derivatives (i.e., clotting factors), disposal of certain surgical instruments, and universal leukoreduction in transfusions. There have been a total of four affected individuals from the United Kingdom who received non-leucoreduced red cell concentrates from UK donors.
This possibility, combined with the probable existence of subclinical carriers, raises the specter of an iatrogenic human-to-human wave of variant CJD transmission. [61] This again highlights the need for reliable detection methods for prion-tainted blood products. [62] In addition, a finding of preclinical infection in a patient heterozygous at codon 129 of PRNP has significant implications regarding the future estimates and surveillance of variant CJD.
In September 2019, the UK Ministers of Health began withdrawal of variant CJD risk reduction measures, including plasma importation and the use of aphoresis platelets, for people born after 1995 or with thombitic thrombocytopenic purpura (TTP). The action was taken on advice from the Advisory Committee on the Safety of Blood, Tissues and Organs (SaBTO). [63]
Epidemiology
A study of 32,441 appendices obtained from 41 hospitals in the United Kingdom, which had been archived over some 6 decades, detected 16 instances of abnormal PrP protein, indicating a prevalence of approximately 1 case in 2,000. There was no bias based on sex, date of collection, or geographical region. Given that the CNS is less hospitable to PrP than lymphoid tissue, this finding does not translate into an assumption of a clinical prevalence of 1 case in 2,000. [64]
According to the World Health Organization fact sheet on variant CJD (revised February 2012), 224 total cases of variant CJD were reported worldwide through March 2011: 175 cases in the United Kingdom and 49 cases in other countries (25 in France, 5 in Spain, 4 in Ireland, 3 in the Netherlands, 3 in the United States, 2 in Canada, 2 in Italy, 2 in Portugal, 1 in Japan, 1 in Saudi Arabia, and 1 in Taiwan). [65, 66, 7] . According to the National CJD Research and Surveillance Unit in Edinburgh, UK (2015), there have been a total of 229 cases of variant CJD reported worldwide and no new-onset cases since 2012.
The vast majority of cases of variant CJD have included documented exposure to food products in countries where bovine spongiform encephalopathy (BSE) occurs, and 2 cases have occurred secondarily as a result of exposure to blood transfusion from individuals who then developed variant CJD. [59, 67] One patient who died of an unrelated cause was found to have a subclinical infection, which was very likely secondary to a blood transfusion from a variant CJD–positive donor who subsequently went on to develop the disease. [60]
Cases of iatrogenic CJD have been identified as resulting from medical treatment, typically from donor blood products obtained from individuals infected with variant CJD. Additionally, corneal grafts, gonadotropin infusion, and contaminated neurosurgical instruments have been responsible for iatrogenic CJD cases. [68]
The number of cases of variant CJD peaked in 2000 in the United Kingdom at 28 and then steadied at 20 cases in 2001, 17 in 2002, and 18 in 2003. Nine new cases were reported in 2004 and another 5 in 2005. The rate has since declined to about 2 cases per year, through 2008. [7] This raises the possibility that the epidemic may have peaked. [69] Despite the optimism, uncertainty remains about the likely size of the total variant CJD epidemic, because such calculations depend on assumptions, including the mean incubation period in humans or the infectious dose of BSE for humans. In contrast, sporadic CJD occurs with a uniform incidence of 1 case per million population per year worldwide. [70] Other forms of prion diseases are even rarer.
Whether the cases from the United Kingdom represent the beginning of an epidemic or whether the numbers will remain low or even continue to decline is unclear. Estimates of the possible size of the epidemic have ranged from 70-136,000 cases. [71, 72] Models provide more conservative estimates of 403-1000 at 95% confidence intervals. [73, 74] It is reassuring that among 63,007 surgically removed tonsils, none tested positive for PrP, [75] as a distinctive PrPSc subtype (4t) is consistently observed in antemortem and postmortem tonsil examinations in cases of variant CJD. [48]
In the United States and Europe, surveillance of patients with prion diseases is performed. Therefore, reporting any suspected prion disease, in particular suspected variant CJD, to surveillance agencies is necessary. Two of the surveillance agencies in the United States are the National Prion Disease Pathology Surveillance Center at Case Western Reserve University in Cleveland, Ohio, and the California Creutzfeldt-Jakob Disease (CJD) Surveillance Project. [33, 76]
Autopsies are performed in only an estimated 22% of cases of CJD in California. The autopsy rates of suspected cases of CJD should be increased because only a pathologic review of tissue can distinguish between classic and variant forms of CJD.
Prognosis
Like other prion-related diseases, variant CJD is relentlessly progressive and inevitably leads to death.
In contrast to the traditional forms of Creutzfeldt-Jakob disease (CJD), variant CJD affects younger patients, with the median age at death being 28 years, as compared with 68 years for traditional forms of CJD. [7] The age range of patients with variant CJD is 14-74 years. [77] Various models suggest that variant CJD infection preferentially affects young people and that older individuals are probably more resistant. [74, 78]
Variant CJD also has a relatively longer duration of illness (median of 14 months for variant CJD, as compared with 4.5 months for traditional CJD). [7]
-
Incidence of bovine spongiform encephalopathy (BSE) and variant Creutzfeldt-Jakob disease (CJD) in Great Britain. The BSE epidemic peaked in 1992, 4 years after the introduction of the ban on ruminant feed. The associated human disease, variant CJD, was not defined until 1996, 7 years after a ban was introduced in Britain on the use of specified offal from cattle in human food.
-
Geographic distribution of bovine spongiform encephalopathy (BSE) by country as of January 9, 2004. From http://www.oie.int/eng/info/en_esb.htm.
-
Time course of epidemic bovine spongiform encephalopathy (BSE) in the United Kingdom, 1986-2000, with dates of major precautionary interventions. SBO stands for specified bovine offal (ie, brain, spinal cord, thymus, spleen, and intestines from cattle aged >6 mo). MBM stands for meat and bone meal (protein residue produced by rendering). From Brown P, Will RG, Bradley R, et al. "Bovine spongiform encephalopathy and variant Creutzfeldt-Jakob disease: background, evolution and current concerns". Emerging Infectious Diseases, 2001;7: 6-16.
-
Normal fluid-attenuated inversion recovery (FLAIR) image at the level of the basal ganglia shows that the thalamus is normally isointense or slightly hypointense relative to putamen. From Collie DA, Summers DM, Sellar RJ, et al. "Diagnosing variant Creutzfeldt-Jakob disease with the Pulvinar sign: MR imaging findings in 86 neuropathologically confirmed cases." Am J Neuroradiol, 2003;24: 1560-9.
-
Pulvinar sign of variant Creutzfeldt-Jakob disease. Fluid-attenuated inversion recovery (FLAIR) image shows marked symmetrical hyperintensity of the pulvinar (posterior) thalamic nuclei, and this sign is present in 100% of cases imaged with FLAIR imaging. From Collie DA, Summers DM, Sellar RJ, et al. "Diagnosing variant Creutzfeldt-Jakob disease with the Pulvinar sign: MR imaging findings in 86 neuropathologically confirmed cases." Am J Neuroradiol, 2003;24: 1560-9.
-
Axial fluid-attenuated inversion recovery (FLAIR) showing periaqueductal gray matter hyperintensity (arrow). Although not a specific sign, periaqueductal hyperintensity is observed in 83% of patients imaged with FLAIR imaging. From Collie DA, Summers DM, Sellar RJ, et al. "Diagnosing variant Creutzfeldt-Jakob disease with the Pulvinar sign: MR imaging findings in 86 neuropathologically confirmed cases." Am J Neuroradiol, 2003;24: 1560-9.
-
Hockey stick sign of variant Creutzfeldt-Jakob disease. Fluid-attenuated inversion recovery (FLAIR) image shows symmetrical pulvinar and dorsomedial thalamic nuclear hyperintensity. This combination produces a characteristic hockey stick appearance and is present in 93% of patients imaged with FLAIR imaging. From Collie DA, Summers DM, Sellar RJ, et al. "Diagnosing variant Creutzfeldt-Jakob disease with the Pulvinar sign: MR imaging findings in 86 neuropathologically confirmed cases." Am J Neuroradiol, 2003;24: 1560-9.
-
Prion protein (PrP) accumulation in the tonsil in variant Creutzfeldt-Jakob disease within follicular dendritic cells and macrophages in a germinal center as demonstrated by PrP immunocytochemistry. From Ironside JW, Frosch MP, Bernardino G. "Human prion diseases." In: Gray F, De Girolami U, Poirier J, eds. Escourelle & Poirier Manual of Basic Neuropathology. Philadelphia, Pa: Elsevier, 2004: 145-57.
-
The florid plaque in the cerebral cortex in variant Creutzfeldt-Jakob disease comprises a dense core with a paler outer layer of amyloid fibrils surrounded by spongiform change (hematoxylin and eosin stain at low magnification). From Ironside JW, Frosch MP, Bernardino G. "Human prion diseases." In: Gray F, De Girolami U, Poirier J, eds. Escourelle & Poirier Manual of Basic Neuropathology. Philadelphia, Pa: Elsevier, 2004: 145-57.
-
The florid plaque in the cerebral cortex in variant Creutzfeldt-Jakob disease comprises a dense core with a paler outer layer of amyloid fibrils surrounded by spongiform change (hematoxylin and eosin stain at high magnification). From Ironside JW, Frosch MP, Bernardino G. "Human prion diseases." In: Gray F, De Girolami U, Poirier J, eds. Escourelle & Poirier Manual of Basic Neuropathology. Philadelphia, Pa: Elsevier, 2004: 145-57.
-
Immunocytochemistry for prion protein (PrP) shows strong staining of the florid plaques and multiple smaller plaques and diffuse PrP deposits (low magnification). From Ironside JW, Frosch MP, Bernardino G. "Human prion diseases." In: Gray F, De Girolami U, Poirier J, eds. Escourelle & Poirier Manual of Basic Neuropathology. Philadelphia, Pa: Elsevier, 2004: 145-57.
-
Immunocytochemistry for prion protein (PrP) shows strong staining of the florid plaques and multiple smaller plaques and diffuse PrP deposits (higher magnification). From Ironside JW, Frosch MP, Bernardino G. "Human prion diseases." In: Gray F, De Girolami U, Poirier J, eds. Escourelle & Poirier Manual of Basic Neuropathology. Philadelphia, Pa: Elsevier, 2004: 145-57.
Tables
Psychiatric Features |
Early Onset < 4 mo |
Later Onset 4 to < 6 mo |
Late Onset =6 mo |
Common (n =50) |
Dysphoria Withdrawal Anxiety Irritability Insomnia Loss of interest |
Poor memory Impaired concentration |
Disorientation Agitation |
Less common (n = 25 to < 50) |
Behavioral changes Anergia Poor performance |
Tearfulness Weight loss Appetite change Hypersomnia Confusion |
Hallucinations Impaired self-care Paranoid delusions Inappropriate affect |
Rare (n < 25) |
Obsessive features Losing things Suicidal ideation Panic attacks |
Psychomotor retardation Diurnal mood variation Loss of confidence |
Bizarre behavior Paranoid ideation Recognition impairment Confabulation Lack of emotion Perseveration Impaired comprehension Change in eating preferences Impaired use of devices Acalculia |
Neurologic Features |
Early Onset < 4 mo |
Later Onset 4 to < 6 mo |
Late Onset =6 mo |
Common (n =50) |
None |
Gait disturbance Slurring of speech |
Hyperreflexia Impaired coordination Myoclonus Incontinence Eye features |
Less common (n = 25 to < 50) |
Pain |
Paresthesia Numbness |
Chorea Extensor plantars Dysphagia Clonus Hypertonia Primitive reflexes |
Rare (n < 25) |
Headaches Dropping things Sweatiness Loss of consciousness |
Tremors Handwriting impairment Coldness Odd sensation Dizziness Cranial motor weakness |
Dysdiadochokinesis Taste disturbance Startle response Hypersensitivity Peripheral motor weakness Primitive reflexes |
Class I |
A - Progressive neuropsychiatric disorder B - Duration of illness longer than 6 months C - Routine investigations not suggestive of alternative diagnosis D - No history of iatrogenic exposure E - No history of familial form of transmissible spongiform encephalopathy (TSE) |
Class II |
A - Early psychiatric symptoms (ie, depression, anxiety, apathy, withdrawal, delusions) B - Persistent painful sensory symptoms (eg, frank pain and/or dysesthesia) C - Ataxia D - Myoclonus or chorea or dystonia E - Dementia |
Class III |
A - EEG without typical appearance of sporadic CJD (ie, generalized triphasic periodic complexes at approximately one per second) or no EEG B - Brain MRI showing bilateral symmetrical pulvinar high-signal intensity (relative to the signal intensity of the other deep gray matter nuclei and cortical gray matter) |
Class IVA |
Positive findings on tonsil biopsy (biopsy not routinely recommended and not recommended in cases with EEG appearance typical of sporadic CJD but may be helpful in suspected cases in which the clinical features are compatible with variant CJD without MRI findings of bilateral pulvinar high-signal intensity) |
Diagnoses |
Definite - Class IA and neuropathological confirmation of variant CJD (spongiform change and extensive prion protein deposition with florid plaques throughout the cerebrum and cerebellum) Probable - Class I, 4 of 5 of class II, class IIIA, and class IIIB; or class I and class IVA Possible - Class I and 4 of 5 of class II and IIIA |




