Factors in Atherosclerosis and Stroke
Evidence continues to accumulate to suggest important roles for inflammation and genetic factors in the process of atherosclerosis and specifically in stroke. According to the current paradigm, atherosclerosis is not a bland cholesterol storage disease, as previously thought, but a dynamic, chronic, inflammatory condition due to a response to endothelial injury. [1]
Traditional risk factors, such as oxidized low-density lipoprotein (LDL) and smoking, contribute to this injury. Infections and inflammatory conditions also contribute to endothelial injury and atherosclerosis. Host genetic factors, moreover, may modify the response to these environmental challenges and are responsible for developmental vascular instability. Inherited risk for stroke is likely multigenic, although specific single-gene disorders with stroke as a component of the phenotype demonstrate the potency of genetics in determining stroke risk.
For further information on hemorrhagic stroke, go to Hemorrhagic Stroke; for more information on ischemic stroke, please see Ischemic Stroke.
For patient education information, see eMedicineHealth's Brain and Nervous System Center as well as Stroke and Stroke-Related Dementia.
Atherogenesis: An Inflammatory Process
Inflammation, endothelial dysfunction, and atherogenesis
Atherogenesis is itself an inflammatory process. Recent evidence suggests that the risk of clinical events is related not only to local factors within the atherosclerotic plaque (such as the state of the necrotic core or the fibrous cap), but also to blood-borne or systemic factors. [1] Thus, circulating levels of cytokines, prothrombotic factors, or acute phase reactants may play a role in precipitating acute stroke in the setting of diseased vessels, even when not stenotic. For example, serum biomarkers, such as high sensitivity C-reactive protein (hsCRP) and cytokine levels, predict progression of atherosclerosis and risk of stroke. [2, 3] Recent infection may also serve as an acute precipitant of vascular events, including stroke. [4]
When endothelium is physically damaged or becomes dysfunctional, a cascade of immunologically-mediated events is precipitated. Potential causes of endothelial dysfunction include sheer stress related to hypertension, oxidized low-density lipoprotein (LDL), homocysteine, and smoking. Dysfunctional endothelium leads to increased permeability to lipoproteins and up-regulation of leukocyte and endothelial adhesion molecules. In response to the presence of certain activating substances, including oxidized LDL, monocyte chemoattractant protein 1 (MCP-1), interleukin (IL)-8, and platelet-derived growth factor (PDGF), leukocytes migrate into the wall of the artery (see the image below).
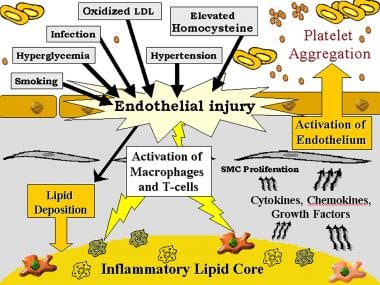 Genetic and inflammatory mechanisms in stroke. Gray background, subendothelium; white background, intraluminal smooth muscle cells (SMC).
Genetic and inflammatory mechanisms in stroke. Gray background, subendothelium; white background, intraluminal smooth muscle cells (SMC).
Induced by oxidized LDL, MCP-1 promotes diapedesis of monocytes across the endothelium. Granulocyte-macrophage colony-stimulating factor transforms monocytes into macrophages, which elaborate tumor necrosis factor (TNF)-α; IL-1; proteolytic enzymes, including matrix metalloproteinases (MMP); and growth factors, including PDGF and insulin-like growth factor (ILGF).
These macrophages, in addition to smooth muscle cells, activate T cells by presenting antigens, including oxidized LDL. Other trophic factors, such as IL-2, TNF-α, and granulocyte-macrophage stimulating factor, cause activated T cells to produce interferon-γ, TNF-α, and TNF-β, leading to stimulation of macrophages and further up-regulation of leukocyte adhesion molecules. Cytokines such as IL-1β and TNF-α are responsible for beginning the response, which is perpetuated by others in the cascade, such as IL-6 and IL-8. This feedback amplifies the cycle of inflammation-mediated events and the release of acute phase reactants (see the image below). [5, 6]
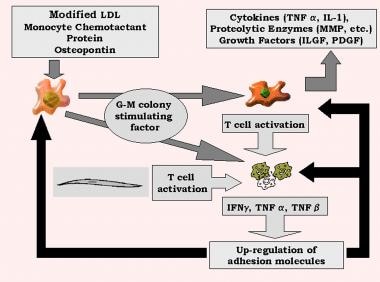 Genetic and inflammatory mechanisms in stroke. G-M, granulocyte-monocyte; IFN, interferon; IL, interleukin; ILGF, insulin-like growth factor; LDL, low-density lipoprotein; MMP, matrix metalloproteinases; PDGF, platelet-derived growth factor; TNF, tumor necrosis factor.
Genetic and inflammatory mechanisms in stroke. G-M, granulocyte-monocyte; IFN, interferon; IL, interleukin; ILGF, insulin-like growth factor; LDL, low-density lipoprotein; MMP, matrix metalloproteinases; PDGF, platelet-derived growth factor; TNF, tumor necrosis factor.
Regulation of adhesion molecules is also influenced by mechanical forces. Low shear stress up-regulates expression of vascular cell adhesion molecule 1 (VCAM-1), while increased shear stress can lead to increased gene expression of intercellular adhesion molecule 1 (ICAM-1). ICAM-1 and VCAM-1 are members of an immunoglobulin superfamily whose members have both a transmembrane region and a cytoplasmic tail. They are expressed on endothelial cells and bind to the integrins CD 11a/CD 18 (lymphocyte function-associated antigen-1) and VLA-4, respectively. CD 11a/CD 18 are found on neutrophils, monocytes, macrophages, and lymphocytes, while VLA-4 is found on monocytes and lymphocytes. [5, 7]
Platelets attach to dysfunctional endothelium, macrophages, and exposed collagen. The activated platelets release granules containing cytokines and growth factors, causing conversion of arachidonic acid to both thromboxane A2, leading to further platelet aggregation, and leukotrienes, thereby amplifying the inflammatory process. Platelets also can be activated by platelet-activating factor (PAF), which is produced by monocytes, endothelial cells, and neutrophils. PAF causes platelet aggregation and degranulation and also can promote leukocyte activation. Next, PDGF, TGF-β, and fibroblast growth factor 2 act to cause smooth muscle cell migration to the site and proliferation. Next, the increased activity of specific chemokines and cytokines (IL-1, TNF-α, PDGF, TGF-β, and osteopontin) leads to formation of a fibrous cap on top of the necrotic core of lipid, leukocytes, and debris.
Plaque rupture—the acute precipitant of approximately 50% of clinical events related to large vessel atherosclerosis—occurs at sites of the fibrous plaque where macrophages stimulated by activated T cells enter and may be encouraged by destruction of the fibrous cap through up-regulation and production of proteolytic enzymes, including collagenases, priming it for ulceration or rupture. [1, 5] The profile of inflammatory cytokines in more advanced lesions, such as those taken from endarterectomy plaque specimens, is predominantly a proinflammatory T cell response. [8]
Inflammation in acute stroke
There is a wealth of information that inflammation contributes to injury after stroke. In stroke models, mice with CD4+ and/or CD8+ T-cell deficiency had better outcomes as measured by improved neurologic function 24 hours after reperfusion, decreased infarct volume, and reduced accumulation of leukocytes and platelets. B-cell deficient mice did not display the same protective effects, implicating a strong T-cell influence. Similarly, studies have shown better outcomes in animals deficient in ICAM-1 and selectin family members. [7, 9, 10]
Elevated levels of MMP-9 have been demonstrated to play an important role in both ischemic stroke and especially hemorrhagic stroke. [11] MMPs have been associated with worse stroke severity, including blood brain barrier dysfunction, increased vascular permeability, larger initial lesion size, growth of infarction, and hemorrhagic transformation. There is also growing evidence suggesting that the hemorrhagic complications of thrombolytic therapy might be mediated by MMP-related activity. [11]
Risk Factors And Mechanisms Of Injury
Infection as a stroke trigger
In light of the increasing acceptance of atherosclerosis as a chronic inflammatory disease, acute infections have been hypothesized to play a role in vascular disease. Acute or recent infections have been particularly well studied as causes of stroke. Several studies provide evidence that patients with stroke are more likely than control subjects to have had an upper respiratory infection within the previous 2 weeks, [12, 13] particularly in large vessel atherothrombotic and cardioembolic cases, even in those without vascular risk factors. [14, 15]
There is a wide range of reported prevalence of infection in ischemic stroke, from 18-40% in the month preceding an event and from 10-35% in the week prior. [4, 14] In a young adult (< 50 y) case series, 35% had a febrile infection in the month preceding an ischemic stroke. [16] A review of case-control studies in adults through 2009 showed a consistent association between ischemic stroke and wide range of exposure to infection 1 week to 1 year prior.
Most often the infection was an upper respiratory tract infection, and the strongest associations were found for younger adult age strata. [17] Both recent upper respiratory and urinary tract infections were associated with increased stroke risk among more than 50,000 stroke patients in the United Kingdom General Practice Research Database; investigators found that patients were more likely to have experienced stroke during the 90 days after these infections. [4]
Using the case-series method, which controls for inter-individual variability, stroke risk in the 3 days after infection was approximately 3 times as high as that during infection-free periods and gradually diminished during the following 3 months.
The association between stroke and preceding infection was also described in children among whom traditional risk factors are often absent. [18, 19] Among children in the Northern Californian Kaiser Pediatric Stroke Study (1993-2003), a minor infection (acute fever, upper respiratory tract infection, pneumonia, acute otitis media, pharyngitis, urinary tract infection, or acute gastroenteritis) independently predicted stroke risk in the following 4 weeks. [20]
Several potential biological mechanisms may explain how infections increase the risk of ischemic stroke. These include increased platelet reactivity, increased leukocyte count, infection-induced autoantibodies, endothelial dysfunction, and atherogenesis. Infections increase platelet reactivity and platelet-leukocyte interactions, leading to an increased risk of platelet aggregation, potentially precipitating stroke. Platelet activation (assessed by P-selectin expression) and platelet-leukocyte aggregates are both increased in stroke patients. [21] In one study, platelet activation and platelet-leukocyte aggregates were also increased among 21 stroke patients with a history of infection within 1 week prior to the stroke, compared with 37 stroke patients without recent infection. Severity of stroke was also greater among those with recent infection.
Other organisms implicated in causing atherosclerosis and ischemic events have also been associated with platelet aggregation, including periodontal infections. [22]
Increased leukocyte count is independently associated in observational studies with carotid [23] and aortic arch plaque thickness, [24] as well as with stroke risk, even after adjusting for smoking. In the Northern Manhattan Study (NOMAS), leukocyte count in healthy elderly individuals was predictive of first ischemic stroke after adjusting for vascular risk factors, including metabolic syndrome and socioeconomic factors. [25] Leukocyte count was also associated with progression of aortic arch atheroma, a risk factor for cardioembolic stroke, over 12 months. [26] Elevated leukocyte count was associated with increased risk of developing a first ischemic stroke or myocardial infarction even after correcting for traditional vascular risk factors. [6]
One of the most convincing analyses of the relationship of leukocyte count to recurrent stroke risk was conducted as a secondary analysis of the Clopidogrel versus Aspirin in Patients at Risk of Ischemic Events (CAPRIE) trial, [27] in which short-term changes in leukocyte count were associated with an increased period of stroke risk. Among 211 patients with ischemic stroke during follow-up, leukocyte levels in the prior week, but not earlier, were increased above baseline levels.
An example of the association of infection-induced autoantibodies with stroke is the heat-shock proteins (HSPs). HSPs are endogenously produced by the arterial wall to protect against hemodynamic-, oxidant-, and cytokine-induced stress. Human HSP60 and mycobacterial HSP65, produced by many bacteria, have high sequence homology. Chronic bacterial infection can lead to cross-reactivity to human HSP60. Antimycobacterial HSP65 has been associated with atherosclerosis. [28, 29] Atherosclerotic disease occurs as a consequence of antibody-mediated vascular endothelial cytotoxicity. [30]
Several chronic or persistent latent infections have been variably associated with atherogenesis and stroke. These agents include Chlamydia pneumoniae, Helicobacter pylori, HIV, herpes viruses, Treponema pallidum, Rickettsia rickettsiae, and Trypanosoma cruzi, among others. At present, no single organism has been consistently shown to be an unequivocal cause of stroke or of atherosclerosis.
If infection is associated with atherosclerosis or stroke risk, it may be that the cumulative inflammatory load from several different infections contributing to this risk. [31] The terms ““pathogen burden” or “infectious burden,” however, have been used to refer to the potential role of multiple infections on an outcome measure such as atherosclerosis. [32]
In a prospective cohort, for example, elevated antibodies to higher numbers of infections, including C pneumoniae, H pylori, Haemophilus influenzae, Mycoplasma pneumoniae, CMV, Epstein-Barr virus, and herpes simplex virus (HSV) types 1 and 2, were more likely to be associated with progression of atherosclerosis. [33] In the NOMAS prospective cohort, a higher burden of antibodies to different agents, including Cpneumoniae, H pylori, CMV, HSV-1, or HSV-2, was associated with maximum carotid plaque thickness and an approximate 40% increased risk of incident stroke. [34, 35]
Chlamydia
C pneumoniae is the infectious agent that has been most extensively studied in relation to atherosclerosis and stroke. C pneumoniae is present in the intima, media, macrophages, and smooth muscle of some carotid endarterectomy specimens. C pneumoniae seropositivity predicted ultrasound-measured atherosclerotic lesion progression in carotid and femoral arteries in the prospective, population-based Bruneck Study. [36] Detection of C pneumoniae in serum, however, correlates poorly with its detection in carotid plaques. [37, 38]
Evidence also exists that patients with coronary disease [39] and stroke [40] are significantly more likely than control subjects to have elevated levels of immunoglobulin G (IgG) or immunoglobulin A (IgA) against C pneumoniae. [41] T-cell proliferation of both CD4+ and CD8+ populations have been demonstrated in C pneumoniae antigen-mediated (positive polymerase chain reaction) symptomatic atherosclerotic plaques. [5, 42] However, prospective studies have not always confirmed these findings. [39]
In animal studies, C pneumoniae inoculation increases atherosclerosis, and azithromycin attenuates this effect. [43] It is unclear whether the organism is causally related to atherosclerosis in humans, however. Large-scale clinical trials have not demonstrated a benefit of antichlamydial therapy in reducing the risk of vascular events in patients with established coronary artery disease. [44, 45, 46, 47] It is possible that other antibiotic regimens or treatment of patients at an earlier stage of disease would still provide benefit.
H pylori
Hpylori chronic infection is highly prevalent, particularly within lower socioeconomic countries. In a series of endarterectomy atheromas, H pylori was found in the majority. [48] A virulent strain, the cytotoxin-associated gene-A (CagA) has been associated with risk of stroke risk [49, 50, 51] and specifically with large-vessel stroke compared with cardioembolic stroke. [52, 51]
Periodontitis
Periodontal disease is another prevalent condition that has been linked to stroke risk. Two periodontal pathogens, Porphyromonas gingivalis and Streptococcus sanguis, have been detected in atherosclerotic plaque. [53] Some periodontal pathogens may invade tissue cultures of both human coronary artery endothelial cells and coronary artery smooth muscle cells. [54] In a meta-analysis of 8 prospective and 1 retrospective cohort studies, periodontal infection generally diagnosed by physical examination indicators was associated with a cardiovascular outcome and independently with risk of stroke. [55] Case-control studies have found a similar association between ischemic stroke patients and examination findings consistent with periodontitis. [56, 57]
Examination findings may not correlate perfectly with periodontitis. A nested case-control study found that serologically confirmed Pgingivalis periodontitis was associated with a higher odds of stroke, [58] but a meta-analysis of studies using periodontitis-specific serology and periodontal bacterial burden showed inconsistent findings for an association with stroke and carotid intimal medial thickness, a measure of atherosclerotic burden. [59] In a randomized study, treatment of periodontitis resulted in early endothelial dysfunction, but improved endothelial function at 6 months, measured through flow-mediated dilatation of the brachial artery. [60]
Herpes viruses
Other chronic viral infections also have been hypothesized to play a pathogenic role in atherosclerosis. Although results have been mixed, some prospective studies found associations between serologic evidence of infection with CMV and HSV and subsequent stroke. [61, 62, 63] CMV has been associated with early and advanced carotid atherosclerosis. [64] In chickens, a herpes virus has been shown to induce atherosclerosis even in normocholesterolemic animals, while vaccines against the virus have been shown to block this response. [65] While the animal data are provocative, human studies have failed to consistently support an association between herpes and atherosclerosis.
Effect of vaccines on stroke
Regarding vaccinations and risk of stroke, there are 2 plausible possibilities: an early increased risk of stroke from immune activation and a protective effect from reduced infectious burden. The UK General Practice Research Database looked at a potential early increased risk of stroke within 91 days of receipt of influenza, tetanus, or pneumococcal vaccine. There was no association with ischemic stroke. [66]
Some evidence from observational studies suggests vaccination against common infections, particularly viral influenza, may prevent stroke. In a case-control study of 370 consecutive stroke or transient ischemic attack patients and an equal number of community controls, influenza vaccination during the previous season was associated with a 50% reduction in stroke risk after adjusting for other risk factors (a 19.2% vaccination rate in patients, compared with a 31.4% rate in control subjects). [67] Using 3 large managed-care databases, vaccination against influenza among older adults (≥65 y) was associated with a 16% and 23% reduction in the risk of hospitalization for stroke. [68] In a randomized intervention study among patients with coronary artery disease, vaccination against influenza resulted in fewer myocardial ischemic events. [69] There are no randomized trials on influenza vaccination and stroke risk.
In children and some adults, varicella infection appears to represent a period of increased stroke risk, and it is possible that vaccination against chickenpox can reduce this risk. [70] Recent guidelines recommend vaccination against influenza in patients with cardiovascular disease. [71]
Influence of infection by stroke subtype
Three case-series studies from single institutions found that large artery atherothrombotic and cardioembolic stroke were both associated with an inflammatory event, including infective and noninfective conditions associated with local or systemic activation of the immune system, within the prior 1-3 months. [72, 73, 74] Among children, the Canadian Pediatric Ischemic Stroke Registry (1992-2008) found that an acute infection at the time of a first cardioembolic stroke was associated with stroke recurrence. [75]
Inflammatory Biomarkers As Predictors Of Stroke Risk
Epidemiological studies have shown that several inflammatory biomarkers are also associated with atherosclerosis and stroke. Even within the normal range, relative increases in these levels may serve as predictors of future risk.
High-sensitivity C-reactive protein (hsCRP) and stroke risk
Acute phase proteins, hsCRP in particular, have been the most extensively studied markers of inflammation. [76] Other markers, including lipoprotein-associated phospholipase A2 (Lp-PLA2), serum amyloid A (SAA), interleukin-6 (IL-6), and CD40 ligand (CD40L), also may predict events. Whether CRP is directly causative of atherosclerosis or simply an epiphenomenon (ie, a marker of the inflammation that is present in atherosclerosis but not directly responsible for it) remains uncertain. Increasing evidence suggests that CRP may play a more direct or causative role or serve as another risk factor for atherosclerosis. In vitro, CRP up-regulates and stimulates the release of several cytokines and growth factors and down-regulates nitric oxide, a potent vasodilator. [77]
HsCRP predicts incident cerebrovascular events in several populations, and much of the early literature on this topic has been reviewed in a European consensus statement. [78] In the Women’s Health Study, hsCRP, IL-6, soluble ICAM-1, and SAA all predicted cardiovascular events, including stroke. [63, 79] In multivariate models, hsCRP was the only inflammatory marker that independently predicted risk, and the inclusion of hsCRP improved the predictive ability of the models over those containing lipid values and other risk factors alone in several studies. [80, 81] These initial studies focused on predominantly healthy middle-aged individuals without a significant burden of risk factors. However, in the Cardiovascular Health Study, which focuses on risk factors in elderly persons, hsCRP predicted a first ischemic stroke only modestly (adjusted hazard ratio for the highest quartile 1.60, 95% confidence interval [CI], 1.23-2.08). [82]
HsCRP levels measured before the onset of clinical disease were an independent predictor of first ischemic stroke (relative risk 1.9 for those in the highest quartile [CRP >2.1] versus those in the lowest quartile) in the Physicians' Health Study, but the effect on stroke risk was less than the effect on cardiac risk and appeared to exhibit a threshold effect above the first quartile. [83] Among those older than 85 years, hsCRP was associated with risk of death from all causes, as well as fatal stroke. [84] In the Framingham Study, men in the highest quartile of hsCRP had twice the risk of stroke of those in the lowest, and women had 3 times the risk. For men, the increased risk was not statistically significant after adjusting for confounders. [85]
Among healthy Japanese American men, baseline hsCRP was associated with increased risk of stroke after 10-15 years of follow-up only among those younger than 55 years, without hypertension, or without diabetes. [86] Thus, hsCRP may be of greatest predictive value among those with low baseline risk and of least value in older populations and those with more risk factors. CRP gene polymorphisms were associated with hsCRP levels, and in white participants, the 1919T allele was associated with an increased risk of stroke and cardiovascular mortality. [87] Thus, the relationship of hsCRP to stroke risk is less certain than its relationship to myocardial infarction, and it appears to depend on study design and population. Validation in other populations is required.
Carotid artery disease
A growing body of literature suggests that inflammatory biomarkers can distinguish symptomatic from asymptomatic carotid plaques and that these markers can be used to predict which patients with carotid stenosis are most likely to develop symptoms. Plaques from symptomatic patients undergoing carotid endarterectomy have significantly more inflammatory cells on immunohistochemical analysis than plaques from asymptomatic patients. [88] Doppler imaging may be a useful way to distinguish stable from unstable carotid plaques, and findings correlate with inflammatory biomarkers.
In one study, plaques in a group of stroke patients were predominantly echolucent, whereas those in the asymptomatic group were predominantly echogenic. [89] Irregular and ulcerated plaques were frequently found in stroke patients, while smooth plaques were frequently detected in asymptomatic patients. Serum levels of matrix metalloproteinase-9, soluble CD40L, and hsCRP were higher in stroke patients than in asymptomatic patients. In the Northern Manhattan Study (NOMAS), irregular and ulcerated plaques were more strongly associated with future stroke risk than smooth plaques. [90]
Some studies have also used high-resolution carotid MRI as an alternative technique to assess the stability of carotid plaque, and visualized abnormalities are associated with levels of serum inflammatory markers. [91] Treatment to lower levels of LDL has also been associated with MRI-visualized carotid plaque regression. [92] Wiart and colleagues demonstrated the potential of using ultrasmall superparamagnetic particles of iron oxide (USPIO) to enhance MRI images in vivo by labeling inflammatory phagocytes of ischemic lesions. [93, 94]
Role of hsCRP for prognosis after stroke
The role of hsCRP and other inflammatory markers as prognostic markers after a first stroke has been investigated in few studies. [78] In a secondary analysis of nested case-control data from a multicenter secondary stroke prevention trial, the Perindopril Protection against Recurrent Stroke Study (PROGRESS), those in the highest tertile of hsCRP had a 40% increased risk of recurrent ischemic stroke. [95] The investigators did not fully adjust for diabetes mellitus. Fibrinogen and hsCRP were associated with increased risk of recurrent ischemic stroke, but not hemorrhagic stroke, after accounting for other risk factors and medications.
A more recent analysis in the same population found that each of these markers was associated with recurrent ischemic stroke. [96] When all markers, including hsCRP, were considered simultaneously, the effect appeared most prominent for TNF-α. The investigators argued that the acute phase response itself, rather than any particular marker, is associated with outcomes.
There is also evidence, perhaps not surprisingly, that hsCRP predicts mortality after stroke. [97, 98] These studies require confirmation in large, multicenter studies with prospectively defined thresholds for marker levels. At present, testing for hsCRP after stroke to predict prognosis or to recommend treatment cannot be routinely recommended. [78]
Lp-PLA2 and stroke risk
Lipoprotein-associated phospholipase A2 (Lp-PLA2) is a macrophage-derived enzyme involved in metabolism of LDL in arterial walls that is responsible for release of inflammatory mediators. [99, 100] Recent epidemiologic studies have provided evidence that relative elevations in serum levels of Lp-PLA2 are associated with increased risk of incident ischemic stroke. [101, 102] This risk appears to be independent of the effect of hsCRP. In NOMAS, hsCRP and LP-PLA2 were assessed as predictors of stroke recurrence, other vascular events, and death among 467 patients with incident stroke. [40] Those in the highest quartile of Lp-PLA2 were at about double the risk of recurrent stroke. They also had an increased risk of the combined outcome of recurrent stroke, myocardial infarction, or vascular death (adjusted HR 1.86, 95% CI, 1.01-3.42). After adjusting for other prognostic factors, hsCRP was not associated with recurrent stroke or other vascular events, but it was associated with mortality.
From this data, it appears that inflammatory markers are associated with prognosis after first ischemic stroke but that different markers offer complementary information. LP-PLA2 may be a stronger predictor of recurrent stroke risk and, therefore, may be more specific for vascular inflammation, while hsCRP, an acute phase reactant, increases with stroke severity, is less specific for vascular inflammation, and is associated with mortality to a greater degree than recurrence. [103]
Potential therapeutic implications
Elevations in inflammatory marker levels could be used to target patients for therapy with secondary prevention medications, including antiplatelets, antihypertensives, and statins. This approach is consistent with the paradigm in primary and secondary prevention, according to which overall baseline level of risk, rather than specific risk factors, should be treated with all drugs efficacious in reducing the risk of vascular events. [25] Several trials demonstrate that hydroxymethylglutaryl-coenzyme A reductase inhibitors or statins reduce levels of hsCRP and Lp-PLA2 independently of effects on cholesterol levels. [104, 105]
Some recent evidence suggests that reduction in hsCRP and Lp-PLA2 may be associated with a reduction in the risk of vascular disease. [106, 107] In the Myocardial Ischemia Reduction with Aggressive Cholesterol Lowering (MIRACL) trial of atorvastatin, hsCRP levels were related to stroke risk over the next 16 weeks. Of note, however, stroke risk was related to hsCRP only in the placebo group, providing indirect evidence that statin treatment can reduce the risk associated with elevated hsCRP. [108]
In the JUPITER trial, which examined the benefits of rosuvastatin therapy in participants with an elevated hsCRP level but low LDL, there was a significant reduction in cardiovascular events, including stroke, in the rosuvastatin-treated group compared with the control patients. [109] The effect appeared independent of lowering LDL. More studies are required to determine what the true extent of the anti-inflammatory role of statin therapy is and whether any modification of current guidelines would be warranted to incorporate measuring additional markers, such as hsCRP. Because the benefit of statins did not seem dependent on hsCRP levels and the limited inclusion criteria, there is still no consensus on what these results mean in terms of primary prevention and treatment.
Because secondary stroke prevention guidelines do not state that all stroke patients should be treated with statin therapy and because stroke is a heterogeneous condition, hsCRP or Lp-PLA2 may be useful as additional markers to help determine which stroke patients should be started on statins. The decision to measure hsCRP, Lp-PLA2, or other markers in stroke patients may be based on the CDC/AHA guidelines until further data are available. [76]
Genetics and Stroke
While the genetic contribution to traditional cardiovascular risk factors, such as hypertension, diabetes mellitus, and high cholesterol, has been thoroughly investigated, the role of genetics in causing stroke itself is beginning to be explored. [110, 111, 112] Several novel genetic determinants of stroke have been discovered in the past few years, in the coagulation pathways, in developmental vascular disorders, and in atherogenesis. The genetics of nontraditional risk factors, including inflammation, have also been increasingly studied in recent years. The effect of exposure to toxins, such as cigarette smoke and infectious agents, also may be modified by genetics. Single-gene stroke disorders, while rare, do occur and provide insight into mechanisms of disease. Selected single-gene disorders are discussed.
Sickle cell anemia
Cerebrovascular complications in sickle cell anemia (SCA) are the result of polymerized red blood cells at low oxygen tensions, resulting in small vessel occlusion and sickle-related arterial disease. SCA is seen in approximately 6% of children with stroke, [113] but one quarter of individuals with SCA experience a stroke by age 45 years and the highest incidence for ischemic stroke is at 2-5 years. [114] By the onset of early adulthood, the risk of ischemic stroke is surpassed by the increased incidence of hemorrhagic stroke. Untreated, the risk of a recurrent stroke is as high as 90%. [115] Multiple silent infarcts may be more common than clinical infarcts, and they can impair cognitive processing and school performance. [116, 117] Sickle-related arterial disease is most often moyamoya disease. A second polymorphism in genes regulating inflammation confers an even higher risk of stroke in these children. [118]
Metabolic, single-gene disorders
Homocystinuria
Homocystinuria is a genetically heterogeneous disease that results in accumulation of homocysteine and premature atherosclerosis. Homocystinuria is well established as a contributor to endothelial damage, proatherosclerotic and thromboembolic generation, lipoprotein oxidation, vascular smooth muscle proliferation, and even arterial dissection. [119, 120] The most common defect is in the enzyme cystathionine beta synthase (CBS), coded for by subband 21q22.3, which normally converts homocysteine to cystathionine.
Methylenetetrahydrofolate reductase (MTHFR) is a folate-dependent enzyme that participates in the remethylation of homocysteine to methionine. Another common mutation, the MTHFR 677C→T polymorphism, has been associated with elevated homocysteine levels and increased risk of stroke. The NOMAS cohort found that total homocysteine levels above 15 µmol/L were independently associated with ischemic stroke. [121] These effects are greatest in populations with low folate intake; however, future trials of folate therapy to reduce homocysteine levels and prevent stroke are needed. [122]
Fabry disease
Fabry disease is an X-linked deficiency (both missense and nonsense mutations) in the lysosomal α-galactosidase A (GLA) gene, resulting in accumulation of globotriaosylceramide in cells throughout the body, including the vascular endothelium. It is the second most common lysosomal storage disorder. Cerebrovascular involvement typically occurs in both large and small vessels and in the posterior circulation, especially of younger stroke patients. [119] Mechanisms of stroke include cardioembolism from cardiac involvement, large artery thromboembolism from dolichoectasia, and tortuosity of large vessels and steno-occlusive disease of the small vessels due to glycolipid accumulation in the endothelium and vascular smooth muscle cells. In the Fabry Outcome Survey, the frequency of stroke among males aged 25-44 years was approximately 12 times the expected frequency in the general population. [123] Enzyme replacement therapy can modify the natural course ofthis disease.
MELAS
The syndrome of mitochondrial encephalopathy, lactic acidosis, and strokelike episodes (MELAS) is a mitochondrial disorder characterized by strokelike episodes often involving the cortex, typically in the occipitoparietal region; migraine headaches; nausea; and vomiting. Multiple organ systems can be affected, leading to short stature, hearing loss, developmental delay, diabetes mellitus, and other problems. Imaging of the vasculature within the lesions reveals no embolic or stenotic mechanisms, but rather a vasogenic impairment of microvascular blood flow autoregulation or metabolism, not limited to particular vascular territories. The significant heterogeneity in the phenotype of family members is thought to arise from heteroplasmy, the variable expression of mutated mitochondrial DNA within different tissues. The most commonly reported genetic defect is an A3243G substitution within a tRNA gene in 80% of cases. [124]
CADASIL
Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) causes granular degeneration of the media of small vessels and a prominent and progressive leukoencephalopathy. The phenotype is characterized by migrainelike headaches, depression and psychosis, and recurrent strokes, often leading to pseudobulbar palsy and subcortical dementia. [125] Characteristic imaging findings include white-matter lesions in the external capsule and anterior temporal poles. [126]
Genetic linkage analyses have identified several associated mutations of the Notch3 gene, which is located on band 19q12. [125] Most of these mutations result from a missense mutation altering the number of cysteine residues expressed in an extracellular receptor domain. [127] Notch3 is widely expressed in the body and plays an important role in development, but the role of Notch3 in normal smooth muscle and why the disease clinically affects only the nervous system is unknown. While the defective receptors usually do not interfere with phenotypic signaling, they have been shown to accumulate in the basilar lamina of arterial vasculature. [128, 129] Skin biopsy revealing granular osmiophilic material can be pathognomonic for the diagnosis, sometimes detecting the disease in patients who have normal findings on imaging studies and negative genetic test results for the most common mutations. [130]
Familial amyloid angiopathy
Mutations in cystatin C have been implicated in familial amyloid angiopathy. [131] Cystatin C mutation is associated with early intracerebral hemorrhages and late degenerative diseases such as Alzheimer disease.
Connective-tissue and basement membrane single-gene disorders
Cerebrovascular complications are commonly associated manifestations of several hereditary connective-tissue diseases. Fibromuscular dysplasia is an example of a noninflammatory, no-atherosclerotic arterial disorder with distinct angiographic findings and a high risk of cerebrovascular events. Among 27 children with pathologically proven fibromuscular dysplasia, 78% had angiographically demonstrated focal stenotic arteriopathy, 63% had ischemic strokes, 37% had hemorrhagic strokes, 40% had multifocal strokes, and 36% had stroke recurrence. [132]
Single-gene disorders, such as Marfan syndrome, Loeys-Dietz syndrome, and Ehlers-Danlos syndrome, caused by mutations in the fibrillin-1 (FBN1), collagen type-III (COL3A1), TGFβ-receptor 1- and -2 genes, and the SMAD3 gene carry a risk of vascular dissections, ischemic infarcts, aneurysms, and cardiovascular disease. There are many single-gene disorders associated with cerebrovascular complications in children and young adults. [133]
COL4A1
Mutations in the COL4A1 gene, encoding the alpha-1 chain of type IV collagen, result in impaired vessel-wall integrity. A range of cerebrovascular manifestations have been described, including small-vessel steno-occlusive disease, aneurysms, and dolichoectasia, with resulting ischemia, intracranial hemorrhage, leukoencephalopathy, and retinal arteriolar tortuosity. [134] An increased tendency for cerebral hemorrhage can occur with head trauma, participation in sports, and anticoagulant use. [135]
ABCC6
Mutations in the ABCC6 gene, important for the assembly and deposition of elastic fibers, results in pseudoxanthoma elasticum (PXE). PXE is characterized pathologically by calcified elastic fibers and phenotypically by peripheral vascular disease, increased skin laxity, redundant skin folds, and papular skin lesions. Neurologically, ischemic strokes, small-vessel disease, aneurysms, and moyamoya disease have been described. This disorder with progressive ectopic mineralization has been described as a metabolic disorder with high morbidity and mortality. [136]
ACTA2
Actin alpha 2 (ACTA2) is an isoform of actin responsible for vascular smooth-muscle contraction. Mutations in the gene lead to alpha-actin polymerization and smooth-muscle cell proliferation. Phenotypically, individuals have evidence of smooth muscle dysfunction throughout the body, including fixed dilated pupils, hypotonic bladder, gut malrotation and hypoperistalsis, and pulmonary hypertension. [137] There is a risk for aortic, cervical, and intracranial arterial dissection. [138] They are also at risk for steno-occlusive small-vessel disease, moyamoya disease, and aneurysmal large-vessel disease. [139]
NF1
Neurofibromatosis type 1 (NF1) is a disorder resulting from a dysfunctional tumor suppressor gene in the Ras signaling pathway. The cerebrovascular expression of disease is excessive smooth cell proliferation and fibrosis with aneurysms and vascular occlusion, such as moyamoya disease. [140] More recent study shows that NF1 in mice with no overt vascular disease have increased circulating monocytes and macrophages in the neointima, suggesting that there may be an inflammatory component to the vascular disease. [141]
ELN
Williams syndrome (William-Beuren syndrome) results from mutation in the ELN gene encoding elastin. The majority of patients have supravalvular aortic stenosis with additional systemic vascular involvement. The elastin deficiency also leads to intimal smooth-muscle cell proliferation. The vascular phenotype is generally occlusive disease and includes moyamoya disease.
JAG1
Alagille syndrome is a multisystem disorder involving the liver, skeleton, heart, eyes, and vasculature with a characteristic facies, the result of mutation in the jagged-1 surface protein (JAG1). JAG1 is involved in notch signaling and vascular development. JAG1 receptors are also involved in the response to cellular injury. Neurologic comorbidity is most commonly secondary to bleeding from aneurysmal rupture. [142]
HTRA1
Cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy (CARASIL) is due to mutations in the HtrA serine peptidase-1 gene. HTRA1 impairment is associated with dysregulation of TGFβ signaling. Patients develop early alopecia and arteriosclerosis with intimal thickening and medial degeneration. Neurologically, there is prominent small-vessel disease and progressive leukoencephalopathy, similar to what is seen with CADASIL. [143]
SLC2A10
Arterial tortuosity syndrome is caused by mutations in SLC2A10 gene encoding a glucose transporter, GLUT10. Pathologically, there is disruption of elastic fibers in the medial layer of the arterial wall with fragmentation of internal elastic membranes. [144] High TGFβ is thought to be a response to unstable elastic membranes. [145] Affected patients have arterial tortuosity, aneurysmal dilations and dissections, and occlusive arteriopathy.
Common ischemic stroke
There is evidence that garden-variety stroke is also associated with familial and, possibly, genetic factors. [146] The identification of genetic factors is still at a nascent stage. Twin studies support heritability, demonstrating a 17.7% concordance rate for monozygotic twins and only 3.6% for dizygotic twins. [147] Genetic influence appears to be more substantial in younger patients. [148, 149] A relationship appears to exist between parental stroke and silent cerebral infarcts in their offspring. The MRI finding of T2 hyperintensities consistent with small vessel ischemic disease in the elderly has concordance rates of 0.61 for monozygotic twins and 0.38 for dizygotic twins, [150] possibly suggesting a genetic susceptibility to a particular stroke mechanism.
One of the first genome-wide single-nucleotide polymorphism (SNP) association studies in ischemic stroke was published in 2007 from 5 US centers. [151] Since that time, several large international stroke cohorts have begun to identify genetic variants even among subtypes of ischemic stroke. In the Wellcome Trust Case Control Consortium 2 (WTCCC2) of UK and German ischemic stroke cohorts, the heritability of previous candidate genes was 37.9% for all ischemic stroke, 40.3% for large-vessel disease, 32.6% for cardioembolic stroke, and only 16.1% for small-vessel disease. [152]
Several genetic variants have been identified. The Northern European multicenter EuroCLOT study evaluates genetic variants in the coagulation and fibrin pathways as risk factors for ischemic stroke. In their first step of a genome-wide association study, they identified 23 statistically independent SNPs in the TwinsUK cohort, with the strongest signals in genes for factors 13, 7, 12, 3, the ABO gene, von Willebrand factor (vWF), and factor 8. [153] There is an association between the ABO blood group and vWF antigen levels. [154, 155]
Unique population-based traits have historically complicated genetic association studies with poor reliability in other populations. Replication studies and international consortia are therefore important. In a Chinese study of susceptibility loci identified in the United States, investigators confirmed the presence of multiple loci in their population. [156] MetaStroke is currently the largest genetic collaboration in ischemic stroke and includes 15 European, North American, and Australian case-control stroke cohorts. Participants are subtyped by ischemic stroke etiology according to a modified TOAST classification system. MetaStroke confirmed an association with ABO gene variants, vWF, and factor 8 among large-vessel and cardioembolic stroke subtypes but not for small-vessel disease. [157]
Subtype-specific genetic variants have been identified. MetaStroke, WTCCC2, an Icelandic stroke registry, and 4 replication cohorts (in Germany, Sweden, and the United Kingdom) showed that the PITX2 and ZFHX3 genes were associated with cardioembolic stroke, and the HDAC9 gene and 9p21 locus were associated with large-vessel stroke. [158, 159, 160] . The Australian Stroke Genetics Collaborative (ASGC) also identified SNPs in 6p21.1 associated with large-artery atherosclerotic stroke. [161]
PITX2 and ZFHX3 have been associated with atrial fibrillation. [162, 163] The WTCCC2 collaboration also identified PHACTR1 in large-vessel disease. [164] Previously, this gene was associated with myocardial infarction. Variation in 9p21 has been strongly associated with coronary artery disease and myocardial infarction in multiple cohorts. [165, 166] In that region, 15 SNPs were evaluated within large stroke samples from South Germany, the United Kingdom, Europe, and North America. Six SNPs were associated with atherosclerotic or large-vessel stroke in adjusted analysis. A population-attributable risk of 20.1% for large-vessel stroke due to SNPs at 9p21 was estimated from the analysis. [167] There are insufficient data on the 6p21.1 locus to understand its role in large-artery atherosclerotic stroke.
From the US prospective Cardiovascular Health Study, 74 SNPs previously associated with coronary heart disease were analyzed in association with incident ischemic stroke. Alleles of the ABCG2 gene, a transporter of sterols, were either protective or deleterious for the risk of incident ischemic stroke among white and black participants. [168]
Considering other definitions of stroke, the Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) consortium has explored genome-wide association studies in MRI-identified silent infarcts and white matter hyperintensities (WMHs). A novel locus at 17q25 was associated with WMHs on MRI. [169] The association between the 17q25 locus and WMH was confirmed in 9 ischemic stroke cohorts from the International Stroke Genetics Consortium, but there was no association between the locus and lacunar stroke, suggesting that the mechanism of risk is likely not through small-vessel disease. [170]
Inherited coagulation disorders have a role in pediatric stroke. A meta-analysis of case-control studies in children showed a higher likelihood for protein C deficiency, antithrombin III deficiency, antiphospholipid antibodies, elevated lipoprotein(a), factor V Leiden, protein S deficiency, factor II G20210A mutation, and MTHFR C677T mutations among children with stroke compared with healthy children. [171] Low activity of ADAMTS13, a cleaving protease for vWF, has also been associated with pediatric stroke. [172] vWF has an important role in hemostasis through bridging platelets to an injured endothelium and as a carrier molecule for factor VIII. vWF deficiency results in a bleeding diathesis, and vWF overproduction results in a thrombophilia tendency.
Other coagulation disorders are not consistently associated with ischemic stroke in adult cohorts. In the Genetics of Early Onset Stroke (GEOS) study, factor V Leiden was not associated with young adult stroke cases compared with controls. [173] Genetic variations in prothrombin, factor V Leiden and MTHFR genes, however, increased the risk of recurrent stroke in young ischemic stroke patients. [174]
Inherited coagulation disequilibrium may also confer risk for adult ischemic stroke. A meta-analysis of polymorphism in endothelium-derived tissue plasminogen activator (TPA) gene among adults with ischemic stroke found that compared with their nonstroke counterparts, the -7351C/T polymorphism in the TPA gene was associated with ischemic stroke and among 3 white studies the polymorphism was associated with large-artery atherosclerosis but not small-vessel disease and cardioembolism. [175] The polymorphism -7351C/T is in the enhancer region of the TPA gene and influences the release of endogenous tPA. [176]
In a large case series from Western Sweden, haplotypes of the fibrinogen gene, independent of fibrinogen level, were associated with ischemic stroke. [177] In other studies, high-normal fibrinogen levels have been associated with ischemic stroke. An earlier meta-analysis showed that within all adult age-categories (40-59 y, 60-69 y, ≥70 y) per 1 g/L increase in fibrinogen level there was an approximate 2-times increase in the hazard of a fatal or nonfatal stroke. [178]
Gretarsdottir et al reported a strong association between gene variations that confer an increased risk of atrial fibrillation and ischemic stroke, especially those originating from cardioembolic sources. [179] Interestingly, there was also a significant association in the noncardiogenic classified events, which the authors speculate is due to the underdiagnosis of atrial fibrillation. One major limitation of the study was that the subjects being studied were primarily of a Northern European-derived population, so it is unknown how generalizable the findings are.
Mutations in genes related to common stroke risk factors, including lipids and hypertension, have been associated with stroke risk. Hepatic lipase has been associated with increased intima media thickness (IMT) and also with risk of stroke in some populations. [180] Variants of the hepatic enzymes microsomal P450 2C9 (CYP2C9) and vitamin K epoxide reductase complex 1 (VKORC1) have been identified to have a significant affect on a patient’s sensitivity to warfarin. [181, 182]
Infection, inflammation, and gene interaction
Genetics play a role in susceptibility to infections. Host defenses are modified by genetics, with certain human leukocyte antigen classes being more associated with autoimmune responses. A ubiquitous infectious agent thus may cause an inflammatory response in some individuals but not in others. This could explain some of the discrepancies in the literature on the association of infectious agents with atherosclerosis. Polymorphisms of the mannose-binding lectin (MBL) gene, which codes for a protein designed to help facilitate phagocytosis, have been shown to increase the risk of infection in humans. These polymorphisms were also found to correlate with the presence and size of carotid atherosclerotic plaques. [183] Similarly, mutations in the IL1ra gene modify the risk of coronary artery disease associated with C pneumoniae infection. [184]
Similarly, the toll-like receptor-4 (TLR4) is expressed on macrophages, endothelial cells, and vascular smooth cells. It binds to bacterial lipopolysaccharide and plays an important role in the innate immune response, and it also binds to other ligands, including modified LDL, fibrinogen, and NF-κB. Ligand binding to this receptor initiates an inflammatory cascade, including the release of cytokines, chemokines, and adhesion molecules. The TLR4 polymorphism Asp299Gly is associated with a reduction in inflammatory mediators, atherosclerosis assessed by noninvasive studies, and the risk of nonfatal myocardial infarction. TLR4 has not been independently associated with ischemic stroke risk. [5, 185, 186, 187, 188] Other TLR haplotypes have been observed with an increased incidence of myocardial infarctions.
An innate susceptibility for an inflammatory response is demonstrated through a major histocompatibility complex class II receptor polymorphism. T cells expressing defective binding sites can be activated in a receptor-independent manner. This mechanism can be triggered by multiple infectious agents, including even Staphylococcus and Streptococcus antigens. [189]
Inflammation-related genes play an important role in atherosclerosis. Oxidized LDL and fatty acids play a critical role in the early stages of atherosclerosis through inflammatory cell adhesiveness to the endothelium and possible binding to scavenger molecules. [190] Among participants in 4 stroke cohorts, common genetic variants in oxidative phosphorylation genes were associated with both ischemic and hemorrhagic stroke. [191]
A haplotype of the promoter region for the glutathione peroxidase gene was associated with an approximate 2-fold increased risk of ischemic stroke in a young adult and a pediatric stroke series. [192] Glutathione peroxidase is an antioxidant with extracellular influence on maintaining bioavailable nitric oxide.
In a meta-analysis of 6 studies including 105 polymorphisms in 64 inflammation- and cardiovascular-related genes, among nonhypertensive participants from 4 white and one Chinese population, lymphotoxin-alpha (LTA) gene polymorphisms were associated with ischemic stroke. LTA induces expression of intercellular adhesion and vascular cell adhesion molecules on endothelial cells. [193] In mice, loss of LTA activity results in smaller atherosclerotic lesions. [194] Haplotypes of the LTA gene thought to enhance transcription were higher among all stroke patients compared with controls, in one sample, and the relative increase in these haplotypes was particularly high among patients with large-vessel ischemic stroke. [195]
A variable number of tandem repeats in the IL-1 receptor antagonist gene was found in nearly a quarter of stroke patients, approximately 3 times more commonly than in healthy controls, in 2 North American cohorts. [196] The IL-1 receptor antagonist functions as an anti-inflammatory cytokine. Point mutation has been associated with coronary atherosclerosis. [197]
Two SNPs in the complement 3 (C3) gene were associated with ischemic stroke in a large Western Swedish case series. [198] C3 is most of the most abundant complement proteins and is pivotal in the activation of the complement system.
Fatty acids are a source of inflammatory mediators, including leukotrienes. The enzyme 5-lipoxygenase (5-LO) participates in the synthesis of leukotrienes and is expressed in vascular tissue. Certain high-risk polymorphisms in the 5-LO gene are associated with increased carotid IMT as well as with increased levels of inflammatory markers. [199] Importantly, the gene may interact with diet, as polyunsaturated n-6 fatty acids are associated with increased IMT, while marine n-3 fatty acids are typically associated with reduced vascular risk, and reduced leukotriene levels are associated with reduced IMT.
Leukotrienes are proinflammatory molecules expressed from macrophages, neutrophils, eosinophils, and mast cells. Genetic variants in the leukotriene pathway, including the 5-lipoxygenase activating protein, leukotriene C4 synthase, and the leukotriene B4 receptor complex, each conferred a higher risk of stroke independently in a UK and German stroke cohort. [200] Although phosphodiesterase 4D was identified early in multiple studies on genetic stroke risk factors, it was found to be only weakly associated with ischemic stroke in the largest meta-analysis to date. [201] It is possible that stronger associations are present in individual populations. There is some indication that genetic variations in the nitric oxide synthase pathway may also be associated with ischemic stroke. [202, 203, 204]
Arachidonate 5-lipoxygenase activating protein (FLAP) is also associated with the risk of stroke in the Icelandic population and some select Northern European populations. [205] FLAP, encoded from the ALOX5AP gene, is an important mediator in the leukotriene activation pathway with roles in atherosclerosis proliferation. There was a 1.8-fold increase in the risk of myocardial infarction and stroke with the Icelandic population. [205] In the Physicians' Health Study cohort, 10 specific Icelandic gene polymorphisms of the ALOX5AP gene were not associated with ischemic stroke. [206]
Cyclooxygenase 2 (COX2) induces production of prostaglandins, which activate MMPs that appear to be important in the destabilization of plaques. Recent evidence suggests that polymorphisms in the gene for COX2 are associated with reduced levels of CRP and risk of both myocardial infarction and stroke. [207] Expression of COX2 and MMPs was significantly lower in atherosclerotic plaques from participants carrying the mutant allele.
A polymorphism in the TNF-α gene, a potent proinflammatory immunomodulator, results in high TNF-α production. Among South Indians, this TNF-α polymorphism was associated with risk of ischemic stroke. [208]
Taken together, these findings provide evidence that genes related to infection and inflammation play an important role in atherosclerosis and risk for ischemic stroke. Further work is needed to confirm many of these findings in other populations. The proinflammatory genotype, resulting in a high cytokine response, may have been an evolutionary advantage. In the past, it likely promoted wound healing and eradication of infections during times of nutritional deficiency. In today's society, with longer life spans, these previously advantageous traits may contribute to atherosclerosis and insulin resistance. In the future, therapies directed at down-regulating or inhibiting inflammation may reduce atherosclerosis and its complications, including stroke.
-
Genetic and inflammatory mechanisms in stroke. Gray background, subendothelium; white background, intraluminal smooth muscle cells (SMC).
-
Genetic and inflammatory mechanisms in stroke. G-M, granulocyte-monocyte; IFN, interferon; IL, interleukin; ILGF, insulin-like growth factor; LDL, low-density lipoprotein; MMP, matrix metalloproteinases; PDGF, platelet-derived growth factor; TNF, tumor necrosis factor.
-
Genetic and inflammatory mechanisms in stroke. The inflammatory cascade.







