Practice Essentials
Perinatal asphyxia, more appropriately known as hypoxic-ischemic encephalopathy (HIE), is characterized by clinical and laboratory evidence of acute or subacute brain injury due to asphyxia. The primary causes of this condition are systemic hypoxemia and/or reduced cerebral blood flow (CBF) (see the image below). Birth asphyxia causes 840,000 or 23% of all neonatal deaths worldwide. [1, 2, 3]
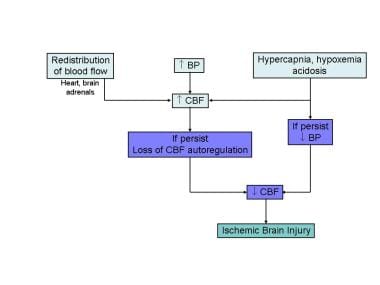 Fetal response to asphyxia illustrating the initial redistribution of blood flow to vital organs. With prolonged hypoxic-ischemic insult and failure of compensatory mechanisms, cerebral blood flow falls, leading to ischemic brain injury.
Fetal response to asphyxia illustrating the initial redistribution of blood flow to vital organs. With prolonged hypoxic-ischemic insult and failure of compensatory mechanisms, cerebral blood flow falls, leading to ischemic brain injury.
Signs and symptoms
Mild hypoxic-ischemic encephalopathy
-
Muscle tone may be slightly increased and deep tendon reflexes may be brisk during the first few days
-
Transient behavioral abnormalities, such as poor feeding, irritability, or excessive crying or sleepiness (typically in an alternating pattern), may be observed
-
Typically resolves in 24h
Moderately severe hypoxic-ischemic encephalopathy
-
The infant is lethargic, with significant hypotonia and diminished deep tendon reflexes
-
The grasping, Moro, and sucking reflexes may be sluggish or absent
-
The infant may experience occasional periods of apnea
-
Seizures typically occur early within the first 24 hours after birth
-
Full recovery within 1-2 weeks is possible and is associated with a better long-term outcome
Severe hypoxic-ischemic encephalopathy
Seizures can be delayed and severe and may be initially resistant to conventional treatments. The seizures are usually generalized, and their frequency may increase during the 24-48 hours after onset, correlating with the phase of reperfusion injury.
As the injury progresses, seizures subside and the electroencephalogram becomes isoelectric or shows a burst suppression pattern. At that time, wakefulness may deteriorate further, and the fontanelle may bulge, suggesting increasing cerebral edema. Other symptoms include the following:
-
Stupor or coma is typical; the infant may not respond to any physical stimulus except the most noxious.
-
Breathing may be irregular, and the infant often requires ventilatory support
-
Generalized hypotonia and depressed deep tendon reflexes are common
-
Neonatal reflexes (eg, sucking, swallowing, grasping, Moro) are absent
-
Disturbances of ocular motion, such as a skewed deviation of the eyes, nystagmus, bobbing, and loss of "doll's eye" (ie, conjugate) movements may be revealed by cranial nerve examination
-
Pupils may be dilated, fixed, or poorly reactive to light
-
Irregularities of heart rate and blood pressure are common during the period of reperfusion injury, as is death from cardiorespiratory failure
An initial period of well-being or mild hypoxic-ischemic encephalopathy may be followed by sudden deterioration, suggesting ongoing brain cell dysfunction, injury, and death; during this period, seizure intensity may increase.
See Clinical Presentation for more detail.
Diagnosis
Guidelines from the American Academy of Pediatrics (AAP) and the American College of Obstetrics and Gynecology (ACOG) for HIE indicate that all of the following must be present for the designation of perinatal asphyxia severe enough to result in acute neurologic injury:
-
Profound metabolic or mixed acidemia (pH < 7) in an umbilical artery blood sample, if obtained
-
Persistence of an Apgar score of 0-3 for longer than 5 minutes
-
Neonatal neurologic sequelae (eg, seizures, coma, hypotonia)
-
Multiple organ involvements (eg, kidney, lungs, liver, heart, intestines)
Laboratory studies
-
Serum electrolyte levels
-
Renal function studies
-
Cardiac and liver enzymes - These values are an adjunct to assess the degree of hypoxic-ischemic injury to the heart and liver
-
Coagulation system - Includes prothrombin time, partial thromboplastin time, and fibrinogen levels
-
Arterial blood gas - Blood gas monitoring is used to assess acid-base status and to avoid hyperoxia and hypoxia, as well as hypercapnia and hypocapnia
Imaging studies
-
Magnetic resonance imaging (MRI) of the brain
-
Cranial ultrasonography
-
Echocardiography
Additional studies
-
Electroencephalography (EEG) - Standard and amplitude-integrated EEG
-
Hearing test - An increased incidence of deafness has been found among infants with hypoxic-ischemic encephalopathy who require assisted ventilation
-
Retinal and ophthalmic examination
See Workup for more detail.
Management
Following initial resuscitation and stabilization, treatment of HIE is largely supportive and should focus on the following [4, 5] :
-
Adequate ventilation
-
Perfusion and blood pressure management - Studies indicate that a mean blood pressure (BP) above 35-40 mm Hg is necessary to avoid decreased cerebral perfusion
-
Careful fluid management
-
Avoidance of hypoglycemia and hyperglycemia
-
Avoidance of hyperthermia - Hyperthermia has been shown to be associated with increased risk of adverse outcomes in neonates with moderate to severe hypoxic-ischemic encephalopathy [6]
-
Treatment of seizures
-
Therapeutic hypothermia (33º-33.5ºC for 72h) followed by slow and controlled rewarming for infants with moderate to severe HIE [7]
See Treatment and Medication for more detail.
Background
Despite major advances in monitoring technology and knowledge of fetal and neonatal pathologies, hypoxic-ischemic encephalopathy (HIE) remains a serious condition that causes significant mortality and long-term morbidity.
HIE is characterized by clinical and laboratory evidence of acute or subacute brain injury due to asphyxia (ie, hypoxia, acidosis). Most often, the exact timing and underlying cause remain unknown.
The American Academy of Pediatrics (AAP) and American College of Obstetrics and Gynecology (ACOG) published guidelines to assist in the diagnosis of severe hypoxic-ischemic encephalopathy (see History). [8, 9]
Pathophysiology
Brain hypoxia and ischemia due to systemic hypoxemia, reduced cerebral blood flow (CBF), or both are the primary physiologic processes that lead to hypoxic-ischemic encephalopathy (HIE). [1, 2, 3]
The initial compensatory adjustment to an asphyxial event is an increase in CBF due to hypoxia and hypercapnia. This is accompanied by a redistribution of cardiac output to essential organs, including the brain, heart, and adrenal glands. A blood pressure (BP) increase due to increased release of epinephrine further enhances this compensatory response. See the image below.
Fetal response to asphyxia illustrating the initial redistribution of blood flow to vital organs. With prolonged hypoxic-ischemic insult and failure of compensatory mechanisms, cerebral blood flow falls, leading to ischemic brain injury.
In adults, CBF is maintained at a constant level despite a wide range in systemic BP. This phenomenon is known as the cerebral autoregulation, which helps maintain cerebral perfusion. The physiologic aspects of CBF autoregulation has been well studied in perinatal and adult experimental animals. In human adults, the BP range at which CBF is maintained is 60-100 mm Hg.
Limited data in the human fetus and the newborn infant suggest that CBF is stable over much narrower range of BPs. [10, 11] Some experts have postulated that, in the healthy term newborn, the BP range at which the CBF autoregulation is maintained may be only between 10-20 mm Hg (compared with the 40 mm Hg range in adults noted above). In addition, the autoregulatory zone may also be set at a lower level, about the midpoint of the normal BP range for the fetus and newborn. However, the precise upper and lower limits of the BP values above and below which the CBF autoregulation is lost remain unknown for the human newborn.
In the fetus and newborn suffering from acute asphyxia, after the early compensatory adjustments fail, the CBF can become pressure-passive, at which time brain perfusion depends on systemic BP. As BP falls, CBF falls below critical levels, and the brain injury secondary to diminished blood supply and a lack of sufficient oxygen occurs. This leads to intracellular energy failure. During the early phases of brain injury, brain temperature drops, and local release of neurotransmitters, such as gamma-aminobutyric acid transaminase (GABA), increase. These changes reduce cerebral oxygen demand, transiently minimizing the impact of asphyxia.
At the cellular level, neuronal injury in HIE is an evolving process. The magnitude of the final neuronal damage depends on the duration and severity of the initial insult, combined with the effects of reperfusion injury, and apoptosis. At the biochemical level, a large cascade of events follow hypoxic-ischemic injury.
Excitatory amino acid (EAA) receptor overactivation plays a critical role in the pathogenesis of neonatal hypoxia-ischemia. During cerebral hypoxia-ischemia, the uptake of glutamate the major excitatory neurotransmitter of the mammalian brain is impaired. This results in high synaptic levels of glutamate and EAA receptor overactivation, including N-methyl-D-aspartate (NMDA), amino-3-hydroxy-5-methyl-4 isoxazole propionate (AMPA), and kainate receptors. NMDA receptors are permeable to Ca++ and Na+, whereas AMPA and kainate receptors are permeable to Na+. Accumulation of Na+ coupled with the failure of energy dependent enzymes such as Na+/ K+ -ATPase leads to rapid cytotoxic edema and necrotic cell death. Activation of NMDA receptor leads to intracellular Ca++ accumulation and further pathologic cascades activation.
EAAs accumulation also contributes to increasing the pace and extent of programmed cell death through secondary Ca++ intake into the nucleus. The pattern of injury seen after hypoxia-ischemia demonstrate regional susceptibility that can be largely explained by the excitatory circuity at this age (putamen, thalamus, perirolandic cerebral cortex). Finally, developing oligodendroglia is uniquely susceptible to hypoxia-ischemia, specifically excitotoxicity and free radical damage. This white matter injury may be the basis for the disruption of long-term learning and memory faculties in infants with hypoxic-ischemic encephalopathy.
Intracellular Ca++ concentration increases following hypoxia-ischemia as a result of (1) NMDA receptor activation, (2) release of Ca++ from intracellular stores (mitochondria and endoplasmic reticulum [ER]), and (3) failure of Ca++ efflux mechanisms. Consequences of increases intracellular Ca++ concentration include activation of phospholipases, endonucleases, proteases, and, in select neurons, nitric oxide synthase (NOS). Activation of phospholipase A2 leads to release of Ca++ from the ER via activation of phospholipase C. Activation of proteases and endonucleases results in cytoskeletal and DNA damage.
During the reperfusion period, free radical production increases due to activation of enzymes such as cyclooxygenase, xanthine oxidase, and lipoxygenase. Free radical damage is further exacerbated in the neonate because of immature antioxidant defenses. Free radicals can lead to lipid peroxidation as well as DNA and protein damage and can trigger apoptosis. Finally, free radicals can combine with nitric oxide (NO) to form peroxynitrite a highly toxic oxidant.
NMDA receptor activation results in activation of neuronal NOS vias PSD-95 and results in the early and transient rise in NO concentration observed in the initial phase of hypoxia. Inducible NOS is expressed in response to the marked inflammation secondary to cerebral ischemia and results in a second wave of NO overproduction that can be prolonged for up to 4-7 days after the insult.
This excessive NO production plays an important role in the pathophysiology of perinatal hypoxic-ischemic brain injury. NO neurotoxicity depends in large part on rapid reaction with superoxide to form peroxynitrite. [12] This, in turn, leads to peroxynitrite-induced neurotoxicity, including lipid peroxidation, protein nitration and oxidation, mitochondrial damage and remodeling, depletion of antioxidant reserve, and DNA damage.
Inflammatory mediators (cytokines and chemokines) have been implicated in the pathogenesis of hypoxic-ischemic encephalopathy and may represent a final common pathway of brain injury. Animal studies suggest that cytokines, particularly interleukin (IL)-1b contributes to hypoxic-ischemic damage. The exact mechanisms and which inflammatory mediators are involved in this process remains unclear.
Following the initial phase of energy failure from the asphyxial injury, cerebral metabolism may recover following reperfusion, only to deteriorate in a secondary energy failure phase. This new phase of neuronal damage, starting at about 6-24 hours after the initial injury, is characterized by mitochondrial dysfunction, and initiation of the apoptotic cascade. This phase has been called the "delayed phase of neuronal injury."
The duration of the delayed phase is not precisely known in the human fetus and newborn but appears to increase over the first 24-48 hours and then start to resolve thereafter. In the human infant, the duration of this phase is correlated with adverse neurodevelopmental outcomes at 1 year and 4 years after insult. [13] See the image below.
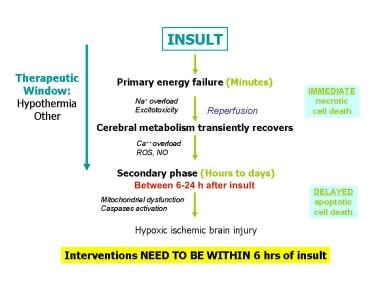 Pathophysiology of hypoxic-ischemic brain injury in the developing brain. During the initial phase of energy failure, glutamate mediated excitotoxicity and Na+/K+ ATPase failure lead to necrotic cell death. After transient recovery of cerebral energy metabolism, a secondary phase of apoptotic neuronal death occurs. ROS = Reactive oxygen species.
Pathophysiology of hypoxic-ischemic brain injury in the developing brain. During the initial phase of energy failure, glutamate mediated excitotoxicity and Na+/K+ ATPase failure lead to necrotic cell death. After transient recovery of cerebral energy metabolism, a secondary phase of apoptotic neuronal death occurs. ROS = Reactive oxygen species.
Additional factors that influence outcome include the nutritional status of the brain, severe intrauterine growth restriction, preexisting brain pathology or developmental defects of the brain, and the frequency and severity of seizure disorder that manifests at an early postnatal age (within hours of birth). [14, 15, 16, 17, 18, 19]
Etiology
Badawi et al investigated risk factors of neonatal encephalopathy in the Western Australian case control study. [20] Of the 164 infants with moderate-to-severe neonatal encephalopathy, preconceptual and antepartum risk factors were identified in 69% of cases; 24% of infants had a combination of antepartum and intrapartum risk factors, whereas only 5% of infants had only intrapartum risk factors. In this study, 5% had no identifiable risk factors. In a review of the literature, Graham et al found that cerebral palsy is associated with intrapartum hypoxia-ischemia in only 14.5% of cases. [21]
Epidemiology
United States data
In the United States and in most technologically advanced countries, the incidence of hypoxic-ischemic encephalopathy (HIE) is 1-4 cases per 1000 births.
International data
The incidence of HIE is reportedly high in countries with limited resources; however, precise figures are not available. Birth asphyxia is the cause of 23% of all neonatal deaths worldwide. It is one of the top 20 leading causes of burden of disease in all age groups (in terms of disability life adjusted years) by the World Health Organization and is the fifth largest cause of death of children younger than 5 years (8%). Although data are limited, birth asphyxia is estimated to account for 920,000 neonatal deaths every year and is associated with another 1.1 million intrapartum stillbirths. More than a million children who survive birth asphyxia develop problems such as cerebral palsy, mental retardation, learning difficulties, and other disabilities. [22, 23]
Prognosis
Accurate prediction of the severity of long-term complications of hypoxic-ischemic encephalopathy (HIE) is difficult, although clinical, laboratory, and imaging criteria have been used. [24] The following criteria have been shown to be the most helpful in outlining likely outcomes:
-
Lack of spontaneous respiratory effort within 20-30 minutes of birth is almost always associated with death.
-
The presence of seizures is an ominous sign. The risk of poor neurologic outcome is distinctly greater in such infants, particularly if seizures occur frequently and are difficult to control.
-
Abnormal clinical neurologic findings persisting beyond the first 7-10 days of life usually indicate poor prognosis. Among these, abnormalities of muscle tone and posture (hypotonia, rigidity, weakness) should be carefully noted.
-
EEG at about 7 days that reveals normal background activity is a good prognostic sign.
-
Persistent feeding difficulties, which generally are due to abnormal tone of the muscles of sucking and swallowing, also suggest significant CNS damage.
-
Poor head growth during the postnatal period and the first year of life is a sensitive finding predicting higher frequency of neurologic deficits.
A Swedish retrospective population-based study comprising 692,428 live births of at least 36 gestational weeks found that more than a quarter (29%) of all HIE births were associated with an obstetric emergency, with parous women affected more than nulliparous women. [130] The investigators noted a strong association of shoulder dystocia in nulliparas, and to uterine rupture in women with previous cesarean deliveries. [130]
Of note, the use of therapeutic hypothermia changes the prognostic value of clinical evaluation in infants with HIE, and its impact on predicting outcomes is still under evaluation. [25]
Other early predictors of long-term neurodevelopmental outcomes are being actively investigated. Early evidence indicates that biomarkers such as serum S100B and neuron-specific enolase may be helpful in identifying infants with severe brain injury who may be candidates for novel neuroprotective or neuroregenerative therapies. [26]
Morbidity/mortality
In severe HIE, the mortality rate is reportedly 25-50%. Most deaths occur in the first days after birth due to multiple organ failure or redirection of care to comfort measures as a result of the grim prognosis. Some infants with severe neurologic disabilities die in their infancy from aspiration pneumonia or systemic infections.
The incidence of long-term complications depends on the severity of HIE. As many as 80% of infants who survive severe HIE develop serious complications, 10-20% develop moderately serious disabilities, and as many as 10% are healthy. Among the infants who survive moderately severe HIE, 30-50% may have serious long-term complications, and 10-20% have minor neurologic morbidities. Infants with mild HIE tend to be free from serious CNS complications.
Two therapeutic hypothermia trials provided updated information on mortality and the incidence of abnormal neurodevelopmental outcomes infants with moderate to severe HIE. [27, 28] In these trials, 23-27% of infants died prior to discharge from the neonatal intensive care unit (NICU), whereas the mortality rate at follow-up 18-22 months later was 37-38%. In these trials, neurodevelopmental outcomes at 18 months were as follows:
-
Mental development index (MDI): Scores of 85 or higher, 40%; 70-84, 21%; less than 70, 39%
-
Psychomotor development index (PDI): Scores of 85 or higher, 55%; 70-84, 10%; less than 70, 35-41%
-
Disabling cerebral palsy - 30%
-
Epilepsy - 16%
-
Blindness - 14-17%
-
Severe hearing impairment - 6%
Data from a randomized controlled trial was evaluated to determine the relationship between hypocarbia and the outcome for neonatal patients with hypoxic-ischemic encephalopathy. The results found that a poor outcome (death/disability at 18-22 mo) was associated with a minimum partial pressure of carbon dioxide (PCO2) and cumulative PCO2 of less than 35 mm Hg; death and disability increased with greater exposure to PCO2 of less than 35 mm Hg. [29]
Even in the absence of obvious neurologic deficits in the newborn period, long-term functional impairments may be present. In a cohort of school-aged children with a history of moderately severe HIE, 15-20% had significant learning difficulties, even in the absence of obvious signs of brain injury. Thus, all children who have moderate or severe HIE should be monitored well into school age. [30, 31, 32]
Race-, sex-, and age-related demographics
No race or sex predilection has been noted.
By definition, HIE is seen in the newborn period. Preterm infants can also suffer from HIE, but the pathology and clinical manifestations are different. Most often, the condition is noted in infants who are term at birth. The symptoms of moderate-to-severe HIE are almost always manifested at birth or within a few hours after birth.
Patient Education
Parents are often concerned about infants' pain and distress, parental-infant bonding, and outcomes following hypothermia treatment. [128] Keys to reassuring parents of infants undergoing hyperthermia include consistent communication, regular updates, and early, balanced discussions regarding potential long-term outcomes; parental involvement in decision making; and having strong support mechanisms. [128]
-
Fetal response to asphyxia illustrating the initial redistribution of blood flow to vital organs. With prolonged hypoxic-ischemic insult and failure of compensatory mechanisms, cerebral blood flow falls, leading to ischemic brain injury.
-
Pathophysiology of hypoxic-ischemic brain injury in the developing brain. During the initial phase of energy failure, glutamate mediated excitotoxicity and Na+/K+ ATPase failure lead to necrotic cell death. After transient recovery of cerebral energy metabolism, a secondary phase of apoptotic neuronal death occurs. ROS = Reactive oxygen species.
-
Severe acute hypoxic-ischemic neuronal change in the basal ganglia is noted. Histologic examination reveals severe hypoxic-ischemic neuronal change, characterized by the presence of pyknotic and hyperchromatic nuclei, the loss of cytoplasmic Nissl substance, and neuronal shrinkage and angulation (arrow). These alterations begin to appear approximately 6 hours following hypoxic-ischemic insult. Reactive astrocytosis is evident approximately 24-48 hours after the primary hypoxic-ischemic event.
-
Significant astrocytosis in the basal ganglia following hypoxic-ischemic insult is observed. An immunohistochemical stain for glial fibrillary acidic protein (GFAP) was performed on the same tissue shown in the previous image to demonstrate the prominent gliosis secondary to the hypoxic-ischemic event. GFAP is a useful marker to study astrocytic response to injury. This gliosis of the basal ganglia, along with subsequent hypermyelination, is responsible for the evolution of status marmoratus over months to years.
-
Bilateral acute infarctions of the frontal lobe are shown. The infarctions depicted in the figure (arrows) are consistent with watershed infarctions secondary to global hypoperfusion. The lesions depicted in the image are consistent with an acute ischemic event, occurring within 24 hours of death. The regions most susceptible to hypoperfusion include the end-artery zones between the anterior, middle, and posterior cerebral arteries.
-
A prior hypoxic-ischemic event involving the occipital lobe has resulted in a chronic lesion marked by dyslamination, neuronal loss, and disorganized arrangements of myelinated white matter fibers. Grossly, the lesion was marked by preserved gyral crests and involved sulci, resulting in prominent, mushroom-shaped gyri.
-
A Luxol-Fast Blue stain was performed on the same tissue shown in the previous image to demonstrate the haphazard arrangement of myelinated white matter fibers projecting into the gray matter of the occipital cortex.
-
Randomized controlled trials of therapeutic hypothermia for moderate-to-severe hypoxic-ischemic encephalopathy (HIE).
-
Periventricular leukomalacia is depicted. This cystic lesion, present in the cingulate cortex, is consistent with periventricular leukomalacia. Note the extensive hemorrhage within the cystic space as well as the hemosiderin-laden macrophages around the lesional rim.
-
Periventricular leukomalacia is depicted. This figure depicts the lesion seen in the previous image at higher magnification. Extensive hemosiderin and reactive astrocytosis is present surrounding the lesion (center of field). Note the proximity of the lesion to the ependymal lining of the lateral ventricle (far right).
-
Summary of potential neuroprotective strategies.
Tables
|
MILD |
MODERATE |
SEVERE |
Level of Consciousness |
Alternating (hyperalert, lethargic,irritable) |
Lethargic or obtunded |
Stuporous |
Neuromuscular Control |
|||
Muscle tone |
Normal |
Hypotonia |
Flaccid |
Posture |
Normal |
Decorticate (arms flexed/legs extended) |
Intermittent decerebration (arms and legs extended) |
Stretch reflexes |
Normal or hyperactive |
Hyperactive or decreased |
Absent |
Segmental myoclonus |
Present |
Present |
Absent |
Complex Reflexes |
|||
Suck |
Weak |
Weak or absent |
Absent |
Moro |
Strong; low threshold |
Weak; incomplete; high threshold |
Absent |
Oculovestibular |
Normal |
Overactive |
Weak or absent |
Tonic neck |
Slight |
Strong |
Absent |
Autonomic Function |
Generalized sympathetic |
Generalized parasympathetic |
Both systems depressed |
Pupils |
Mydriasis |
Miosis |
Variable; often unequal; poor light reflex |
Heart Rate |
Tachycardia |
Bradycardia |
Variable |
Bronchial and Salivary Secretions |
Sparse |
Profuse |
Variable |
GI Motility |
Normal or decreased |
Increased; diarrhea |
Variable |
Seizures |
None |
Common; focal or multifocal |
Delayed |
EEG Findings |
Normal (awake) |
Early: low-voltage continuous delta and theta Later: periodic pattern (awake) Seizures: focal 1-to 1-Hz spike-and-wave |
Early: periodic pattern with Isopotential phases Later: totally isopotential |
Duration |
1-3 days Typically < 24h |
2-14 days |
Hours to weeks |

What would you like to print?
- Overview
- Presentation
- DDx
- Workup
- Treatment
- Medical Care
- Initial Resuscitation and Stabilization
- Supportive Care in Patients with Hypoxic-ischemic Encephalopathy
- Perfusion and Blood Pressure Management
- Fluid and Electrolytes Management
- Hyperthermia Avoidance
- Treatment of Seizures
- Hypothermia Therapy
- Future Neuroprotective Strategies
- Consultations
- Diet
- Prevention
- Long-Term Monitoring
- Show All
- Medication
- Questions & Answers
- Media Gallery
- Tables
- References






