Overview
Background
Disorders of carbohydrate metabolism occur in many forms. The most common disorders are acquired. Acquired or secondary derangements in carbohydrate metabolism, such as diabetic ketoacidosis, hyperosmolar coma, and hypoglycemia, all affect the central nervous system. Many forms and variants of peripheral nerve disease also are seen in diabetes. The remaining disorders of carbohydrate metabolism are the rare inborn errors of metabolism (ie, genetic defects).
The inherited defects affecting carbohydrate metabolism that have been discovered so far are inherited as autosomal recessive traits. Although multiple affected siblings may be identified in a kindred, most affected individuals are the first identified in a family. Most of the known defects of carbohydrate metabolism appear to be due to point mutations. Whether this will still hold true after the discoveries that the Human Genome Project and CRISPR gene editing will make possible is not known.
Most of the inherited disorders of carbohydrate metabolism fall into a few broad clinical syndromes, which are classified by age of onset as follows:
-
Infants and early childhood
Episodic lactic acidosis from early infancy, failure to thrive, and hypotonia with or without features that may suggest specific defects
Infantile or early childhood intellectual disability, hypotonia, failure to thrive, and other features
Intellectual disability/developmental delay with features that suggest storage disorders
Episodic vomiting in infants or young children
-
Childhood or adolescence
Episodic acidosis in childhood or early adulthood, often with features of a specific disorder
Intermittent or episodic ataxia in childhood and adolescence
-
In adults and, more rarely, in adolescents or older children
Cramps and weakness, often with some episodes of muscle breakdown and myoglobinuria, during or after heavy anaerobic exercise
Symmetrical neuromuscular disease with weakness and wasting of proximal muscles
Pathophysiological Basis of Acquired Disorders
The brain, heart, skeletal muscle, and liver depend on ketone bodies or on glucose for energy and for carbon chains to synthesize cellular proteins (see the images below). For infants as well as individuals with decreased sugar intake because of starvation or dietary choices, ketones are the preferred fuel and source of carbon chains. Nervous and muscle tissues preferentially oxidize ketones over fatty acids, glucose, or amino acids under normal conditions. [1, 2, 3, 4]

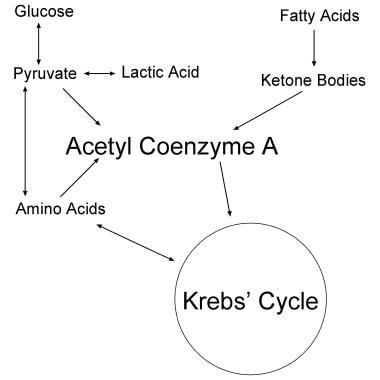
Ketone bodies are largely beta-hydroxybutyrate and acetoacetate. These are formed in the liver and kidneys from the breakdown of fatty acids and a few amino acids. The brains of adults who live on a high-carbohydrate diet lose the ability to synthesize the key enzymes needed to metabolize ketones. These include beta-hydroxybutyrate dehydrogenase and acetoacetyl coenzyme A (CoA) synthetase (see the image below). For the brain to resynthesize these enzymes to effective levels, starvation for 3-7 days or a high-fat diet is needed.
Neural tissue has far more metabolic activity than other tissues of the body. The brain comprises only a small percentage of total body weight—approximately 1.4 kg in an average 70-kg person—but uses 20% of the body's oxygen at rest. In addition, neurons (and glia) are constantly synthesizing a variety of neurotransmitters, proteins for axonal flow, and proteins and lipids for regeneration of synaptic vesicles and other components of membranes. Many of these chemicals are synthesized in the brain, in part or wholly from glucose or ketone bodies.
Nonglucose carbohydrates (eg, galactose, mannose, inositol) are clinically important in fetal and neonatal nutrition; however, little is known about their metabolism in the neonate. Glucose and galactose increase postprandially and several other carbohydrates contained in milk do not. Brown et al, in their attempt to determine whether postprandial changes in plasma carbohydrate and sugar alcohol concentrations are affected by clinical variables such as postnatal age, milk type, feeding volume, or feeding duration in term newborns, found that galactose is almost cleared completely by the neonatal liver. This provides novel evidence of a marked difference in plasma galactose concentrations [Gal] between relatively younger and older neonates. [5]
Acute hypoglycemia, as in an alcoholic or a diabetic who has taken too much insulin, causes the brain to be deprived rapidly and abruptly of blood glucose after it has depended for a long time on glucose rather than ketones as its fuel. Beta-hydroxybutyrate dehydrogenase and acetoacetyl CoA synthetase activity in the brain is insufficient to enable the blood ketones to sustain the brain's metabolism. Without fuel to burn, the neural tissue cannot carry out the high level of metabolism needed to pump ions, fire messages, or transmit intercellular chemical messages, let alone repair itself. The groggy state of sudden memory loss (ie, disorientation) and other intellectual deficits that internists tend to call confusion sets in rapidly with acute hypoglycemia, followed just as rapidly by a downhill course into obtundation, coma, and death. If the average adult brain can withstand only 4-6 minutes of oxygen deprivation, it can withstand only about 40-45 minutes without fuel.
Most physicians, as medical students or interns, have had experience treating diabetic ketoacidosis. Although the experience tends to cast ketone bodies as toxins, they are not. The acidosis produced by ketone bodies is toxic and is the direct cause of the Kussmaul breathing typical of patients with diabetic ketoacidosis. Patients with diabetes in this state usually have had normal to elevated blood glucose for the previous days or weeks. Enough glucose enters the brain cells during this time to inhibit synthesis of beta-hydroxybutyrate dehydrogenase and acetoacetyl CoA synthetase.
Again, the ketone-utilizing enzymes in the brain are not working at effective levels. By the time frank ketoacidosis sets in, too little insulin is left for the blood glucose to be able to sustain the high metabolic activity of the brain. The ketone bodies accumulate but cannot provide energy or carbon for the brain. The hydrogen ion associated with the ketones also accumulates, thereby stimulating (or overstimulating) acid receptors in the floor of the fourth ventricle and interfering with reuptake of potassium in the renal tubules.
The coma of hyperosmolar diabetic disorder seems to be related directly to the addition of large concentrations of glucose in the serum, the cerebrospinal fluid (CSF), the extracellular fluid of the brain and, perhaps to some degree, the intracellular fluid as well. The cause of high concentrations of glucose in these patients but not of ketone bodies remains unknown. The lack of ketones probably explains the lack of acidosis. CSF concentrations of glucose mirror those of the serum with a delay of approximately 2 hours. Patients do not regain consciousness immediately when the blood glucose is lowered.
Pathophysiology of Inherited Disorders
Many of the clinical features of the inherited disorders of carbohydrate metabolism are caused by the following:
-
Lack of glucose for the metabolism of brain, muscle, liver, or kidney (in circumstances in which ketone bodies cannot be used)
-
Inability to break down glucose to pyruvate
-
Inability to oxidize pyruvate fully in the Krebs cycle
Some features of the inherited diseases are due to excessive storage of abnormal substrates or to normal substrates that cells cannot degrade normally because they lack a specific enzymatic activity. In some diseases, abnormal metabolites (intermediates of pathways that use carbohydrates) block the normal function of the pathway or of related pathways. Defects of the enzymes of the pentose shunt interfere with the normal production of nucleic acids, which are needed by cells as second messengers and as coenzymes of intermediary metabolism, as well as components of RNA and DNA.
Galactose accumulates in the lens of the eye in galactose kinase deficiency (see the image below for a review of galactose metabolism).
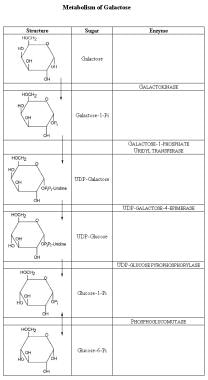 Metabolism of galactose. The galactose ring differs from glucose in that the hydroxyl group (OH) at carbon-3 is up and not down. To convert galactose to glucose, the ring is first bound to uridine diphosphate (UDP). In this form, the sugar moiety can be epimerized, putting the OH down and forming glucose. The UDP-glucose then is converted to glucose-1-phosphate, then to glucose-6-phosphate, and subsequently metabolized further.
Metabolism of galactose. The galactose ring differs from glucose in that the hydroxyl group (OH) at carbon-3 is up and not down. To convert galactose to glucose, the ring is first bound to uridine diphosphate (UDP). In this form, the sugar moiety can be epimerized, putting the OH down and forming glucose. The UDP-glucose then is converted to glucose-1-phosphate, then to glucose-6-phosphate, and subsequently metabolized further.
The osmotic pressure generated causes water to enter the lens; this disruption leads to cataract formation. A similar mechanism may play a role in the pathophysiology of diabetic peripheral neuropathy, which was at one time thought to be due, in part, to the accumulation of sorbitol, a relatively inert but osmotically active metabolite of glucose, and hence to excess water in the cells of peripheral nerves.
See the images below for the major biochemical pathways involved in carbohydrate metabolism.
Metabolism of galactose. The galactose ring differs from glucose in that the hydroxyl group (OH) at carbon-3 is up and not down. To convert galactose to glucose, the ring is first bound to uridine diphosphate (UDP). In this form, the sugar moiety can be epimerized, putting the OH down and forming glucose. The UDP-glucose then is converted to glucose-1-phosphate, then to glucose-6-phosphate, and subsequently metabolized further.
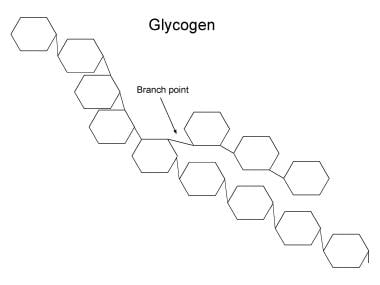 Structure of glycogen. The rings each indicate a glucose moiety in glycogen. These are linked by an ether bond between carbon-1 of one moiety and carbon-4 of the next. In the hydrolysis of glycogen, glycogen phosphorylase (ie, phosphorylase) catalyses the hydrolysis of the glucose moiety from the nonreducing end of glycogen to glucose-1-phosphate and a glycogen chain 1 unit shorter in length than the original. Phosphoglucomutase converts the glucose-1-phosphate to glucose-6-phosphate. Glucose-1-phosphate can then be metabolized, eg, by glycolysis. The branches of glycogen comprise 1-6 ether links. These have to be broken by the glycogen debranching enzyme. In fact, the debranching enzyme has to act before phosphorylase can hydrolyze moieties within 5 units of a branch point. In defects of phosphorylase, glycogen cannot be broken down in the tissue lacking the enzyme; therefore, glycogen accumulates, eventually in very large amounts. In defects of the debrancher, the breakdown of glycogen stops when 5 glucose units still are present near the branch point in any direction. Short branches of glycogen accumulate.
Structure of glycogen. The rings each indicate a glucose moiety in glycogen. These are linked by an ether bond between carbon-1 of one moiety and carbon-4 of the next. In the hydrolysis of glycogen, glycogen phosphorylase (ie, phosphorylase) catalyses the hydrolysis of the glucose moiety from the nonreducing end of glycogen to glucose-1-phosphate and a glycogen chain 1 unit shorter in length than the original. Phosphoglucomutase converts the glucose-1-phosphate to glucose-6-phosphate. Glucose-1-phosphate can then be metabolized, eg, by glycolysis. The branches of glycogen comprise 1-6 ether links. These have to be broken by the glycogen debranching enzyme. In fact, the debranching enzyme has to act before phosphorylase can hydrolyze moieties within 5 units of a branch point. In defects of phosphorylase, glycogen cannot be broken down in the tissue lacking the enzyme; therefore, glycogen accumulates, eventually in very large amounts. In defects of the debrancher, the breakdown of glycogen stops when 5 glucose units still are present near the branch point in any direction. Short branches of glycogen accumulate.
 Glycolysis. The names of sugars are given with a "-" to separate the phosphate moiety so as to emphasize the name of the sugar at each step. As the first step in glycolysis, glucose must enter the cell. Entry is highly dependent on insulin and the reactions at and adjacent to the cell membrane, which are induced by insulin. Next, glucose is given a phosphate handle, so the enzymes of glycolysis (and those of the pentose shunt, glycogen synthesis, further metabolic steps) can handle glucose. When glucose comes from glycogen, it is in the form of glucose-1-phosphate, which has to be converted to glucose-6-phosphate. The glucose moiety is isomerized to fructose, and a second phosphate handle is added to the 6-carbon chain. The chain is then split into two 3-carbon units, each with a phosphate handle. Only one of the 2 forms of P-3-carbon can be metabolized further. The inert form, dihydroxyacetone phosphate, is converted into the active form via the enzyme triosephosphate isomerase. Two molecules of ATP are used up in giving the 6-carbon chain its 2 handles. Nicotine adenine diphosphate (NAD+) is reduced to NADH in the dehydrogenase reaction. In pure glycolysis, these are restored in the oxidative reactions that convert the triose phosphates to pyruvate and the reduction of pyruvate to lactic acid. At the same time, since 2 3-carbon moieties are oxidized for each single 6-carbon moiety that goes down the pathway, 2 extra ATPs are formed. Pure glycolysis thus can generate energy anaerobically. In aerobic metabolism, the bulk of the pyruvate is converted to acetyl coenzyme A rather than to lactate.
Glycolysis. The names of sugars are given with a "-" to separate the phosphate moiety so as to emphasize the name of the sugar at each step. As the first step in glycolysis, glucose must enter the cell. Entry is highly dependent on insulin and the reactions at and adjacent to the cell membrane, which are induced by insulin. Next, glucose is given a phosphate handle, so the enzymes of glycolysis (and those of the pentose shunt, glycogen synthesis, further metabolic steps) can handle glucose. When glucose comes from glycogen, it is in the form of glucose-1-phosphate, which has to be converted to glucose-6-phosphate. The glucose moiety is isomerized to fructose, and a second phosphate handle is added to the 6-carbon chain. The chain is then split into two 3-carbon units, each with a phosphate handle. Only one of the 2 forms of P-3-carbon can be metabolized further. The inert form, dihydroxyacetone phosphate, is converted into the active form via the enzyme triosephosphate isomerase. Two molecules of ATP are used up in giving the 6-carbon chain its 2 handles. Nicotine adenine diphosphate (NAD+) is reduced to NADH in the dehydrogenase reaction. In pure glycolysis, these are restored in the oxidative reactions that convert the triose phosphates to pyruvate and the reduction of pyruvate to lactic acid. At the same time, since 2 3-carbon moieties are oxidized for each single 6-carbon moiety that goes down the pathway, 2 extra ATPs are formed. Pure glycolysis thus can generate energy anaerobically. In aerobic metabolism, the bulk of the pyruvate is converted to acetyl coenzyme A rather than to lactate.
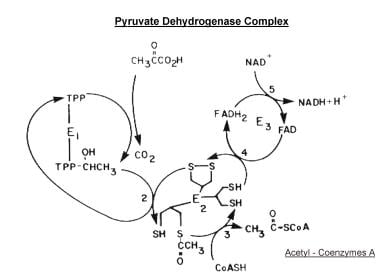 Scheme of the major reactions of the pyruvate dehydrogenase complex. Initial oxidative decarboxylation of pyruvate is shown as reaction (1), transfer of the 2-carbon moiety to the lipoyl side chain of lipoyl acetyltransferase (E2) as reaction (2), formation of acetyl coenzyme A as reaction (3), oxidation of the lipoyl moiety as reaction (4), and reduction of NAD as reaction (5). The regulatory enzymes acting on pyruvate decarboxylase (E1) are not shown.
Scheme of the major reactions of the pyruvate dehydrogenase complex. Initial oxidative decarboxylation of pyruvate is shown as reaction (1), transfer of the 2-carbon moiety to the lipoyl side chain of lipoyl acetyltransferase (E2) as reaction (2), formation of acetyl coenzyme A as reaction (3), oxidation of the lipoyl moiety as reaction (4), and reduction of NAD as reaction (5). The regulatory enzymes acting on pyruvate decarboxylase (E1) are not shown.
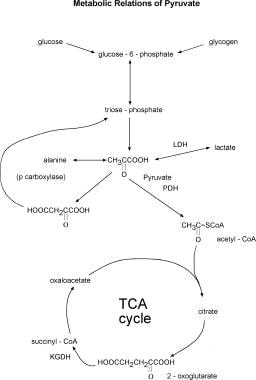 Major metabolic relations of pyruvate. Many of the reactions, such as those between glucose-6-phosphate and pyruvate, pyruvate and alanine, and pyruvate and lactate, are of course reversible. Abbreviations: LDH - lactic dehydrogenase; PDH - pyruvate dehydrogenase complex; p carboxylase - pyruvate carboxylase; TCA cycle - Krebs tricarboxylic acid cycle; KGDH-a-ketoglutarate dehydrogenase complex.
Major metabolic relations of pyruvate. Many of the reactions, such as those between glucose-6-phosphate and pyruvate, pyruvate and alanine, and pyruvate and lactate, are of course reversible. Abbreviations: LDH - lactic dehydrogenase; PDH - pyruvate dehydrogenase complex; p carboxylase - pyruvate carboxylase; TCA cycle - Krebs tricarboxylic acid cycle; KGDH-a-ketoglutarate dehydrogenase complex.
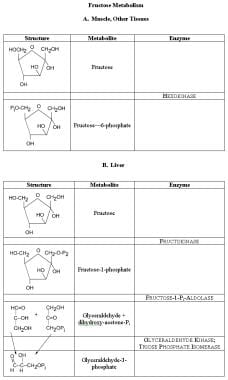 Fructose metabolism. In muscle and most tissue, the same hexokinase that phosphorylates glucose to glucose-6-phosphate also phosphorylates fructose to fructose-6-phosphate. Fructose-6- phosphate then undergoes metabolism, down to pyruvate or up to glycogen, through the enzymes of glycolysis. In the liver, fructose is phosphorylated largely to the 1-phosphate form and converted to glyceraldehydes and dihydroxyacetone phosphate. Free glyceraldehydes can be phosphorylated by a specific kinase; this and dihydroxyacetone are metabolized further by the reactions of glycolysis.
Fructose metabolism. In muscle and most tissue, the same hexokinase that phosphorylates glucose to glucose-6-phosphate also phosphorylates fructose to fructose-6-phosphate. Fructose-6- phosphate then undergoes metabolism, down to pyruvate or up to glycogen, through the enzymes of glycolysis. In the liver, fructose is phosphorylated largely to the 1-phosphate form and converted to glyceraldehydes and dihydroxyacetone phosphate. Free glyceraldehydes can be phosphorylated by a specific kinase; this and dihydroxyacetone are metabolized further by the reactions of glycolysis.
The expression of a disease often depends on other factors in addition to the causative agent. In inherited disease, variations in the other 30,000 genes, varying diets, infectious disease, access to medical care, and even upbringing and cultural effects can influence the clinical expression of a disease. Intermittent ataxia of childhood from pyruvate decarboxylase deficiency tends to appear in conjunction with certain infections or emotional stress, just as is the case with the neurological manifestations of phytanic acid storage disease (Refsum disease).
Patterns of Occurrence
Frequency
The acquired disorders of carbohydrate metabolism are fairly common, both in the United States and internationally. Hypoglycemia is a common cause of neurological disease, especially acute mental deterioration, memory loss, disorientation, obtundation, and coma, among both alcoholics and patients with diabetes who are treated with insulin. [6] Hyperinsulinemia from other causes is rare, but pancreatic tumors could be the cause. Diabetes, with its various neurological complications, is among the most common disorders treated in adult patients. Diabetic ketoacidosis still occurs, though education and close medical follow-up make it less common than it was several decades ago. Hyperosmolar coma is also less a problem than when it was first brought to the attention of internists by Plum and Posner's classic monograph Diagnosis of Stupor and Coma. [7] Hyperosmolar coma still occurs and needs to be kept in mind while evaluating an obtunded patient.
The inherited disorders of carbohydrate metabolism are rare. Severe defects of the pyruvate dehydrogenase (PDH) complex and the benign chemical anomaly called pentosuria have been reported in very few (2-6) patients. Patients with inherited disorders of carbohydrate metabolism tend to be seen at tertiary centers, where specialist teams are available. The team may comprise pediatricians, pediatric neurologists, biochemical geneticists, psychiatrists, neurologists with a special interest in intellectual disability, and neurologists interested in neuromuscular diseases and the more unusual ataxias.
Neurologists in general practice see few, if any, cases of inherited disorders of carbohydrate metabolism in their careers. This depends partly on whether the practice is in an area in which the carrier state for the inherited disorders is more common and partly on whether the neurologist is on the alert for these disorders. As in every area of medicine, inherited carbohydrate disorders that are looked for tend to be found and those that are not looked for tend to be overlooked. Although each of these disorders was described between 1930 and 1990, certainly they were present in the populations of medically sophisticated countries decades, perhaps centuries, earlier.
Glass et al performed a retrospective chart review of new pediatric patients seen during 1998 by specialists of the Division of Clinical and Metabolic Genetics of the Hospital for Sick Children in Toronto, the largest such referral center in the country. They found that 81% of specific genetic metabolic diagnoses were made within 1 month of being seen in consultation. Four years after initial consultation, a specific diagnosis was not reached in 5% of patients. The authors concluded that the specific diagnosis of inborn errors of metabolism at a major medical genetic referral center tended to be made quickly, or never. Some of the causes of delays in diagnosis include the lack of ready access to existing diagnostic laboratory testing, technical barriers to the identification of specific metabolic or genetic defects, and incomplete knowledge of genetic defects causing inherited metabolic diseases. [8]
Mortality
Hypoglycemia, diabetic ketoacidosis, and hyperosmolar coma are all potentially fatal but potentially curable conditions. Mortality depends on the time taken by the patient or patient's family to present medical attention, the time taken by the doctor to consider the diagnosis and begin correct treatment, the thoroughness of the treatment, and the time taken to diagnose and thoroughly treat other associated medical problems. The patient with acidosis benefits very little if the ketosis is corrected but an underlying cause is missed, for example, by a delayed spinal tap if meningitis or encephalitis happens to be the illness that has tipped the metabolic balance.
Some of the inherited disorders of carbohydrate metabolism are fatal. For some of these conditions, the more profoundly the mutation interferes with enzyme activity, the more likely the disorder will be fatal. Other inherited disorders usually are not fatal if treated early and thoughtfully. Pentosuria is not even a disease in the strict sense; it is more a metabolic curiosity without significant clinical consequences.
In general, patients with inherited carbohydrate disorders that primarily affect the brain or heart tend to die early; those whose disorders affect the liver and kidney die later on; and those whose disorders affect only muscle have lower mortality rates, unless things go very wrong. Some concern exists among physicians who study heat illness in military recruits that subclinical inherited predispositions to disorders of muscle and of RBCs might be among the factors that predispose to heat illness. As heat illness is the major cause of death of military recruits in boot camp, this is an issue that deserves careful study.
Race
No racial predilection has been reported in most inherited disorders of carbohydrate metabolism. One well-known exception is pentosuria, a very rare genetic occurrence of no clinical significance, which is so far confined to Ashkenazi Jews. The pentose excreted in this condition is xylulose.
Sex
No sex predilection has been reported; the known inherited disorders of carbohydrate metabolism are all autosomal recessive.
Age
A general rule exists in inherited diseases. If the degree of deficiency of an enzyme varies from family to family, symptoms are likely to appear earliest in families in which the mutation virtually inactivates the enzyme activity and latest in families in which the activity is only just below a certain minimum symptomatic threshold.
Profound defects of activity of the PDH complex are associated with lactic acidosis, hypotonia, profound intellectual disability, and often death in the first months or first year of life. Somewhat milder defects and vigorous treatment can be compatible with survival into early to mid childhood.
Moderate defects of the PDH complex are associated with ataxia and other symptoms and signs presenting in childhood and compatible with graduation from college and survival at least into the adult years. Data from studies in the 1970s and 1980s raised the question whether mild defects of this complex might be associated with brain diseases whose onset is in adolescence or early adult life and that are compatible with long survival. These studies have not yet been refuted or disproved.
Diagnosis and Treatment of Acquired Disorders
Initial management
Diabetic ketoacidosis, hyperosmolar coma, and hypoglycemia present as obtundation or coma. The differential diagnosis is as extensive as it is for obtundation and coma in general. The first clinical task is to make sure the patient survives. Checking the airway, breathing, and circulation (ABCs) and correcting any problems with these is always the first step. The second step is to get baseline blood levels for later analysis before metabolically altering the system during treatment. The third step is to give intramuscular (IM) thiamine followed by 50 mL of 50% intravenous (IV) glucose.
Hypoglycemia can be fatal if not reversed in approximately 45 minutes. A glucose load depletes thiamine stores and precipitates Wernicke syndrome; therefore, thiamine should be given first. Then perform a rapid general examination, looking for clues of serious trouble (eg, abnormal patterns of breathing, unusual body or breath odor, jaundice, pink skin, pallor, petechiae, meningismus, abnormalities of major organs). Next, perform a neurological examination focused on variable conditions associated with coma. Fisher, Plum, and Posner have written extensively about the neurological examination of comatose patients.
The neurological examination helps clarify whether the problem is focal or systemic (metabolic). If all signs point to a lesion on one side or in one location, the lesion is focal; imaging studies and IV steroids need to be considered and a neurosurgeon consulted. If the signs are diffuse or do not lateralize, the problem most likely is metabolic. Again, Fisher and Plum and Posner have published schemata that can be followed to go through the myriad possibilities in a logical manner. Subtle clues from the history or the examination can be crucial and therefore must be given serious consideration and addressed immediately.
Bhattacharya et al, in their pilot study compared uncooked cornstarch with uncooked physically modified cornstarch (WMHM20) in patients with glycogen storage disease types Ia, Ib, and III. The authors reported longer duration of euglycemia and better short-term metabolic control in most of the patients with WMHM20 when compared with cornstarch. [9]
Diabetes
The acute severe disorders in patients with diabetes are diabetic ketoacidosis (see Diabetic Ketoacidosis) and diabetic hyperosmolar coma (see Hyperosmolar Coma). Hypoglycemia from lack of liver glycogen stores (as in alcoholics) or from excess insulin, whether accidental or intentional, also is an acute emergency. The chronic disorders are more common. These include mononeuritis from diabetes, multiple neuropathy (still called mononeuritis multiplex by some) in patients with diabetes, polyneuropathy in patients with diabetes, and so-called diabetic amyotrophy.
Diabetic ketoacidosis
Even though the diagnosis may be established clinically, and even though someone experienced in the care of this problem may sometimes treat the ketoacidosis successfully without supporting laboratory data, confirming the diagnosis by lab studies is still important. Whether the patient will respond easily or with difficulty or what hazards and problems may lie ahead cannot be known at the outset. The extent of Kussmaul breathing is a guide to the severity of acidosis; sudden relief of this breathing, as the patient is being treated, usually means the pH of the blood is back to normal. This can be checked with serial measurements of arterial blood pH (ie, blood gases) while treating the patient.
Ketones and sugars can be demonstrated in the urine and blood by dipstick, but with each repeated step of treatment, sending samples of blood for an accurate analysis by the laboratory is advisable. Changes on the ECG may suggest hypokalemia, but, again, getting the laboratory's ongoing help with serial assays of electrolytes, BUN, and blood pH is advisable. The workup and treatment must include any likely coincidental problem (ie, a new illness other than diabetes) that has tipped the metabolic balance.
Diabetic hyperosmolar coma and hypoglycemia
For more information, refer to Hypoglycemia. These are the 2 major diagnoses to consider in any patient presenting with obtundation or coma of metabolic rather than structural etiology. Once the comatose patient's ABCs have been secured, several tubes of blood must be drawn for the host of cellular and biochemical analyses a comatose patient may need. Administer 100 mg thiamine IM and then inject an IV bolus of 50 mL of a 50% solution of glucose through a needle of largest possible diameter (the author uses a 15-gauge needle). This amount of glucose is enough to reverse hypoglycemia but represents a mere "drop in the bucket" for a patient with hyperosmolar coma or even diabetic ketoacidosis.
Rapid clinical improvement after a bolus of glucose confirms the diagnosis of hypoglycemia. Patients with hyperosmolar coma do not respond, but the initial blood glucose, as measured in the laboratory, is very high (ie, usually well over 400 mg/dL), as is the serum osmolality. The CSF glucose corresponds to the blood glucose but with a 2-hour delay; therefore, the CSF glucose remains high even after the blood glucose and electrolytes are brought slowly back to normal. Remember the dangers of central pontine myelinolysis when correcting electrolyte imbalances due to any cause.
The chronic neurological complications of diabetes are due to damage to peripheral nerves. Mononeuritis and multiple neuropathy are both due to infarction of the tiny artery supplying a particular nerve, the vasa nervorum. Diabetes is a common cause of these problems, and it often presents with infarction of a single nerve, especially of cranial nerves III and VI and of the long mixed nerves of the limbs. When the distal motor nerves of limbs are involved, the diagnosis may be fairly clear. When the proximal motor nerves are primarily involved, the diagnosis may be difficult. The pattern can be that of weakness of the muscles of one thigh with deep pain (ie, diabetic amyotrophy).
Amyotrophy can be a presentation of diabetes. Sometimes, the opposite limb is affected much later. Amyotrophy tends to be self-limiting and improves without any treatment after a few months. The cause of the waxing and waning of symptoms of amyotrophy is unclear.
In some patients, the pattern is of damage to the lumbar plexus, resulting in diabetic plexitis. Again, the reason for the differences in the extent of involvement is unknown.
The differential diagnosis of mononeuritis includes local pressure along the course of the nerve or its roots from trauma, hematoma, entrapment, abscess, or growths, and other causes of infarction of the arteria nervorum, of which a current leading cause is collagen-vascular disease, especially the peculiar form of periarteritis nodosa that is confined largely to the peripheral nervous system (ie, not clinically affecting other organ systems). These may be suggested by repeated abnormalities of the erythrocyte sedimentation rate or the C-reactive protein or by consistent elevations of antibodies specific for certain collagen-vascular diseases.
Other causes of multiple neuropathy include Lyme disease, sarcoidosis, acquired and inherited amyloidosis, and, very rarely, first attacks of tumacular neuropathy (for more information, refer to Lyme Disease and Sarcoidosis and Neuropathy). Most cases of multiple neuropathies are treatable; therefore, this syndrome is one of the few clinical indications for a nerve biopsy should it be needed to establish the diagnosis. The sural nerve is almost always the nerve of choice. Consult an experienced surgeon to obtain the specimen. Too small a piece, or one that looked liked nerve but was collagen, does no good, and the patient usually will not consent to a second attempt at biopsy.
Many cases of plexitis, whether brachial or lumbar, unilateral or bilateral, are due to infectious or postinfectious problems. These need to be excluded carefully before diabetes is accepted as the cause of plexitis. The problem is complicated by treatment. For example, the nondiabetic causes of plexitis often respond to high-dose steroids. Most of the author's patients have responded well to pulse-dose steroids for 5 days as currently used for other immune-mediated neurological disorders. Steroids elevate blood sugars. This means that diabetes needs to be sought and excluded before a therapeutic trial and that a patient with diabetes who is to be given even pulse-dose steroids needs careful monitoring, often with testing of blood glucose 4 times a day for monitoring excessive elevations of glucose during treatment.
In the era before CT scans and MRI of the abdomen and pelvis were available, the worsening of what appeared to be a lumbar plexitis with high-dose steroids pointed to the correct problem, pressure palsy from a pelvic abscess.
Polyneuropathy
Diffuse disorders of peripheral nerves exist, both sensory and motor, but often the sensory disorders are the most profound, clinically affecting the longest fibers first. This is a common problem in neurology in general, and it is also a common problem in people with diabetes. The patients have numbness and tingling in the toes and feet and, later, up the legs. By the time the sensory loss reaches the knees, the fingertips begin to be involved. Often, small-fiber sensation (eg, superficial touch, pinprick, appreciation of hot and cold) is involved early and most predominantly. These patients are likely to scald themselves by getting into too hot a bathtub. They test the water with their feet, but their feet can no longer warn them of the high temperature.
To some extent, the severity of the neuropathy correlates with the severity of the diabetes and with episodes of lack of metabolic control. To some extent, the neuropathy may improve if control is restored. However, the correlation is not at all tight. Some patients presenting with neuropathy have mild diabetes; in others, the neuropathy occurs and does not get better readily, regardless of the degree of control of blood glucose levels. Some patients present with a marked dysesthetic pain, others do not. In many patients, later rather than sooner, a marked autonomic component is present, which is not present in others. When the autonomic system is involved, the distal skin often becomes mottled, cyanotic or pink, smooth, and loses hair follicles to become shiny. Capillary refill may become poor. Postural hypotension occurs in advanced stages of autonomic involvement.
Treatment of complications of the peripheral nervous system in a patient with diabetes is still poor. Good control of blood glucose levels helps prevent or delay onset of these complications; therefore, young patients today often are put on insulin pumps, and blood glucose is monitored much more closely.
The neuropathic pain of diabetes often responds to modern antiepilepsy drugs, as does neuropathic pain of other etiologies, including gabapentin (Neurontin), carbamazepine (Tegretol), and oxcarbazepine (Trileptal). Botulinum toxin-A (Botox) has been used for painful diabetic neuropathy. [10]
Diagnosis and Treatment of Inherited Disorders
Episodic lactic acidosis in infancy, failure to thrive, and hypotonia with or without features suggesting specific disorders
In this category, an infant presents with signs of acidosis, tachypnea, marked irritability, and tachycardia. As the acidosis worsens, the infant becomes obtunded or comatose. Muscle tone is very poor; the infant is flaccid and has poor head control. If the infant is more than a few hours or days old, signs of failure to thrive may be seen: weight loss or at least poor gain in weight, microcephaly, and poor linear growth. Prognosis is poor even with current vigorous metabolic management. Without such management, the infant usually dies in a short time.
In the absence of other clinical features, this may be the presentation of one of several defects of the PDH multienzyme complex, particularly of its E components (see the image below).
Scheme of the major reactions of the pyruvate dehydrogenase complex. Initial oxidative decarboxylation of pyruvate is shown as reaction (1), transfer of the 2-carbon moiety to the lipoyl side chain of lipoyl acetyltransferase (E2) as reaction (2), formation of acetyl coenzyme A as reaction (3), oxidation of the lipoyl moiety as reaction (4), and reduction of NAD as reaction (5). The regulatory enzymes acting on pyruvate decarboxylase (E1) are not shown.
The components are called E1 (ie, pyruvate decarboxylase [and the phosphorylase and dephosphorylase that modify the activity of E1]), E2 (ie, lipoyl acetyl transferase), and E3 (ie, lipoamide, also known as dehydrogenase; dihydrolipoamide: NADH oxidoreductase).
Several cases of Leigh syndrome appear to have been caused by failure of activation of pyruvate decarboxylase, presumably because of defects in phosphorylation. (Three of the 4 infants described thus far with defects of E3 also had the complex clinical picture described by Leigh. [11] ) In theory, a profound congenital deficiency of thiamine ought to produce episodic lactic acidosis in infants with failure to thrive and hypotonia. This picture also can be seen in infants suffering from deficiencies of glucose-6-phosphatase or phosphoenolpyruvate carboxykinase; some infants with the latter also have hepatomegaly.
Leigh syndrome (ie, subacute necrotizing encephalomyelopathy) usually begins in the first or second year of life. Motor and cognitive regression occur along with ataxia, chorea, and tonic spasms. Depending on the age of the patient, history and physical findings include loss of muscle tone and head control, decreased appetite, nausea, vomiting (often with lactic acidosis), irritability to the point of continuous crying, myoclonus, and primarily generalized seizures. Episodes of hyperventilation, apnea and gasps, sobbing without tears, external ophthalmoplegia with pendular or rolling nystagmoid movements of the eyeballs, loss of swallowing, muscle wasting, and loss of reflexes are observed.
In cases of postnatal lactic acidosis with elevations in the levels of ammonia, citrulline, and lysine, consider defects of the B-complex of pyruvate carboxylase, the first enzyme in the pathway that generates glucose from fatty acids and amino acids through gluconeogenesis (see the image below). Death occurs by about 3 months of age.
Major metabolic relations of pyruvate. Many of the reactions, such as those between glucose-6-phosphate and pyruvate, pyruvate and alanine, and pyruvate and lactate, are of course reversible. Abbreviations: LDH - lactic dehydrogenase; PDH - pyruvate dehydrogenase complex; p carboxylase - pyruvate carboxylase; TCA cycle - Krebs tricarboxylic acid cycle; KGDH-a-ketoglutarate dehydrogenase complex.
If the infant vomits shortly after meals and has jaundice, a bleeding diathesis, hepatomegaly, or renal tubular acidosis, check the blood glucose and urinary reducing substances. If blood glucose is low and the reducing substances positive, the infant may have hereditary fructose intolerance. Ascertain whether fructose in the milk or in the formula is in fact producing the vomiting. If tachypnea alternates with apnea, the patient has hepatomegaly, and ketones and lactic acid are present in the blood or urine, consider fructose 1, 6-diphosphatase deficiency (see the image below). Both disorders can be rapidly fatal if untreated and may be fatal even with optimal medical management.
Fructose metabolism. In muscle and most tissue, the same hexokinase that phosphorylates glucose to glucose-6-phosphate also phosphorylates fructose to fructose-6-phosphate. Fructose-6- phosphate then undergoes metabolism, down to pyruvate or up to glycogen, through the enzymes of glycolysis. In the liver, fructose is phosphorylated largely to the 1-phosphate form and converted to glyceraldehydes and dihydroxyacetone phosphate. Free glyceraldehydes can be phosphorylated by a specific kinase; this and dihydroxyacetone are metabolized further by the reactions of glycolysis.
Differential diagnosis
Lactic acidosis in infants is a serious emergency. The vast majority of cases have an acquired cause. These include IV glucose given to a newborn; shock, even hypoxia, from different causes; infectious and parainfectious causes (eg, sepsis even without shock, bacterial meningitis, urinary tract infection with the lactate-producing bacterium Enterobacter cloacae, Reye syndrome); poisoning by salicylates; seizures (seizures lead to increase in CNS lactic acid that results in increase in systemic lactic acid as in patients with bacterial meningitis); liver failure; and short gut syndrome.
After all of these have been ruled out, the inherited disorders need to be excluded, including organic acidemia (especially glutaric acid), disorders of metabolism of branched chain amino acids (especially the biotin-dependent carboxylases), disorders of the PDH multienzyme complex (eg, E1, E2, E3, pyruvate decarboxylase kinase [activator], PDH phosphatase), as well as disorders of gluconeogenesis caused by disruption of the metabolic pathways used to break down glycogen (eg, glucose-6-phosphatase deficiency) and acetyl-CoA (eg, pyruvate carboxylase deficiency, fructose 1, 6-diphosphatase deficiency).
Workup
The initial workup readily identifies lactic acidosis. Once the more common acquired causes of lactic acidosis have been excluded, order appropriate tests to diagnose one of the less common inborn errors of metabolism. Assays for urine organic acids and plasma amino acids need to be performed for specific enzymes when clinically indicated. Initiate the assay up front for an infant with a sibling whose history is consistent with acute lactic acidosis. For the most part, specific enzyme assays can be performed with cultured cells such as skin fibroblasts or on blood lymphocytes, but checking on laboratory specifications for each enzyme in question is important. Robinson has written an excellent scholarly review of the inherited causes of lactic acidosis, their diagnosis, and their treatment. [12] Concise reviews about the workup of lactic acidosis in infants and children also can be found in several recent textbooks of child neurology.
Thiamine can be assayed directly (eg, through Specialty Laboratories of Santa Monica, California). Also, thiamine can be assayed indirectly by assaying RBC transketolase and reassaying after adding excess thiamine pyrophosphate to the test tubes. Several commercial laboratories can perform this assay.
PDH multienzyme complex or its components are difficult to assess. The assays are even more difficult in the tiny amounts of tissue available from cultured or biopsied material from patients. The assays require a research laboratory, such as the ones at the Hospital for Sick Children in Toronto, the Institute of Neurology in New York City, and the laboratory at 785 Mamaroneck Avenue, White Plains, NY.
Infants and young children with von Gierke disease (ie, glucose-6-phosphatase deficiency) have low blood sugars and elevated serum lactic acid, elevated uric acid (if the child is young), and often extremely high triglycerides. Neither glucagon nor catecholamines correct the hypoglycemia in these patients because they lack glycogen stores; instead, lactic acid rises. The definitive diagnosis depends on assay of the enzyme activity or immune cross-reactivity in biopsy specimens of the liver or kidney.
Phosphoenolpyruvate carboxykinase deficiency gives rise to episodic lactic acidosis and low blood glucose. Phosphoenolpyruvate carboxykinase exists in 2 forms, one in the cytosol and the other in mitochondria. Defects of the latter can be shown in cultured fibroblasts, but defects of the former have to be demonstrated in samples of liver.
Pyruvate carboxylase deficiency leads to elevations of ammonia, citrulline, and lysine, in addition to lactic acidosis. This is a biotin-requiring enzyme. It is present in mitochondria and can be assayed in any tissue with mitochondria, including cultured tissues.
Diagnosing defects of fructose 1, 6-diphosphatase deficiency by direct enzyme assay on biopsies of liver or jejunum or on carefully handled leukocytes or cultured tissues is preferable to a fructose challenge that can induce a metabolic crisis.
Treatment
Treatment is difficult. Introduction of the appropriate gene for the normal enzyme into the cells of the body, including the brain, appears to be the desired approach. As yet, this cannot be done safely and routinely, although active research on ways to do it has been going on for 20 years. Acidosis and low blood glucose need to be corrected vigorously with IV glucose and bicarbonate. Robinson suggests combinations of low-carbohydrate or ketogenic diets, thiamine, carnitine, and/or dichloroacetate for patients with defects of the PDH complex. He suggests high-carbohydrate diets for defects of glucose-6-phosphatase, fructose 1, 6-diphosphatase, and pyruvate carboxylase. [12] Note that if a ketogenic diet is used, it needs to be started slowly and pushed only until ketones are consistently present in the urine and not to the point of inducing acidosis, which is sometimes the end point when ketogenic diets areusedtotreat childhood epilepsies.
Avoid all forms and sources of fructose in patients with any of the defects of fructose metabolism, severe or mild.
Plum and his colleagues have shown that lactic acid destroys astrocytes. [13] Since these cells carry nutrients from the blood to neurons and also remove a number of neurotransmitters from synaptic clefts, their loss means the loss of neurons. Treating lactic acidosis is urgent but often difficult. The current methods are to give IV bicarbonate in adequate quantities to normalize the blood pH and, at the same time, to try to stop or reverse the process leading to the production of excess lactate (often as the by-product of excess pyruvate production) in the brain. The treatment of lactic acidosis can require heroic measures. Cederbaum and colleagues reported giving several liters of bicarbonate in one night to a 20-pound infant with severe deficiency of PDH, E1. [14] Decreasing glycolysis may take ingenuity or may be nearly impossible.
Parenthetically, the observation that lactic acid kills glia is one of the reasons many neurologists no longer use glucocorticosteroids routinely in patients with an acute stroke. It would be very interesting to see if the penumbra of strokes would be smaller, on the whole, if patients were treated with IV solutions of ketones, amino acids, or even lipids instead of with solutions of dextrose or lactate. The proposed solutions would need to be used in the ambulance and the emergency department, not merely in the ICU or neurological ward.
Infants with intellectual disability, hypotonia, failure to thrive, and other features
If the infant has survived the first weeks or months of life and begins to show signs of profound intellectual disability, other diagnoses must be considered. Episodic tachypnea interspersed with apnea may suggest lactic acidosis, especially if triggered by fasting or fever. Poor muscle tone, a moderately enlarged liver with low blood glucose, and ketosis indicate deficiency of fructose 1, 6-diphosphatase. In the presence of a more or less permanent metabolic acidosis, poor muscle tone, and myoclonic jerks or frank seizures, look for glycine in the urine by chromatography (not by enzymatic assay). The diagnosis may be D-glyceric aciduria. The enzymatic assays only show L-isomers of amino acids.
Infants with inanition, failure to thrive, intellectual disability, and episodic vomiting (especially after feedings) may have a galactose metabolism problem. Look for cataracts, hepatomegaly, jaundice, elevated liver enzymes, and hyperammonemia. Deficiencies of galactose-1-phosphate uridylyl transferase and of uridine diphosphate galactose-4-epimerase both have been associated with this clinical picture (see the image below).
Metabolism of galactose. The galactose ring differs from glucose in that the hydroxyl group (OH) at carbon-3 is up and not down. To convert galactose to glucose, the ring is first bound to uridine diphosphate (UDP). In this form, the sugar moiety can be epimerized, putting the OH down and forming glucose. The UDP-glucose then is converted to glucose-1-phosphate, then to glucose-6-phosphate, and subsequently metabolized further.
Differential diagnosis
Infantile or early childhood intellectual disability, hypotonia, and failure to thrive, with or without other features, describe the clinical presentations of a large number of disorders.
Workup
In addition to the enzymes listed above, consider the following:
The presence of glycine in the urine by chromatography (not by enzyme assay) is suggestive of D-glyceric aciduria. D-glycine does not react in the enzyme assay because the enzyme is specific for the L-isomer.
If clinical evidence suggests a defect of galactose metabolism, episodes of vomiting follow feedings of milk or formula containing lactose; if galactose levels in the blood or urine are elevated, look for liver damage (ie, elevated ammonia) or renal damage (ie, proteinuria, which may occur 1-2 days after a lactose load and then disappear). The RBCs may have very high levels of galactose-1-phosphate. Galactosemia due to galactose-1-phosphate uridylyl transferase and uridine diphosphate galactose-4-epimerase deficiencies give similar clinical and biochemical pictures. Both enzymes can be assayed directly in the RBCs. A benign variant of uridine diphosphate galactose-4-epimerase deficiency is characterized by deficiency of the enzyme only in the RBCs; enzyme levels are normal in other tissues. This is presumably due to different isoenzymes being present in different tissues.
Mild degrees of galactose kinase deficiency present solely with cataracts in childhood. This enzyme also can be assayed in RBCs.
The mainstay of treatment of individuals with galactosemia due to galactose-1-phosphate uridylyl transferase deficiency has been elimination of lactose (ie, milk sugar) from the diet. Since galactose is present in a number of fruits as well as milk products, certain fruits also should be avoided in order to reasonably eliminate galactose from the diet. However, despite early detection and optimal dietary treatment, children and adults with galactosemia are still at risk for cognitive deficits, and galactosemic women are also at risk for ovarian dysfunction.
Women who have given birth to a child with this form of galactosemia (and are therefore at 25% risk of having another affected child) often are advised to maintain a galactose-restricted diet in subsequent pregnancies. This recommendation is made because evidence exists to suggest that exposed fetuses may be at risk of developing cataracts prior to birth and that some cognitive deficits may be mitigated by such maternal dietary restriction. See the image below.
Metabolism of galactose. The galactose ring differs from glucose in that the hydroxyl group (OH) at carbon-3 is up and not down. To convert galactose to glucose, the ring is first bound to uridine diphosphate (UDP). In this form, the sugar moiety can be epimerized, putting the OH down and forming glucose. The UDP-glucose then is converted to glucose-1-phosphate, then to glucose-6-phosphate, and subsequently metabolized further.
The basis for treatment of other disorders of galactose metabolism is to avoid galactose from all sources, including lactose and foods containing the simple sugars.
Intellectual disability/developmental delay with features that suggest storage disorders
The features that suggest storage disorders are hepatomegaly, a large forehead with frontal bossing, and macrocephaly. A large number of diseases, including storage disorders caused by inborn errors of metabolism, can cause hepatomegaly in infancy without acidosis or other specific features discussed. Of the disorders of carbohydrate metabolism, these features can be seen with defects of production, storage, or catabolism of glycogen, especially deficiency of the glycogen debrancher enzyme, which leads to hepatomegaly in infancy and early death; defects of 3 of the 4 proteins that make up phosphorylase kinase; and deficiency of glucose-6-phosphatase (ie, von Gierke disease), which is accompanied by gout, lactic acidosis, and elevated lipids. Also, a similar clinical picture is seen with the lysosomal disorder of glycogen, Pompe disease. Patients with these disorders tend to have low blood glucose levels.
Differential diagnosis and workup
Intellectual disabilityn/developmental delay with features that suggest storage disorders are described in the same sources as the disorders already discussed and in other articles on lysosomal disorders, disorders of lipid metabolism, and disorders of mucopolysaccharide metabolism in Medscape Reference Neurology. [15]
These features also suggest defects in the production, storage, or catabolism of glycogen (see the images below).
Structure of glycogen. The rings each indicate a glucose moiety in glycogen. These are linked by an ether bond between carbon-1 of one moiety and carbon-4 of the next. In the hydrolysis of glycogen, glycogen phosphorylase (ie, phosphorylase) catalyses the hydrolysis of the glucose moiety from the nonreducing end of glycogen to glucose-1-phosphate and a glycogen chain 1 unit shorter in length than the original. Phosphoglucomutase converts the glucose-1-phosphate to glucose-6-phosphate. Glucose-1-phosphate can then be metabolized, eg, by glycolysis. The branches of glycogen comprise 1-6 ether links. These have to be broken by the glycogen debranching enzyme. In fact, the debranching enzyme has to act before phosphorylase can hydrolyze moieties within 5 units of a branch point. In defects of phosphorylase, glycogen cannot be broken down in the tissue lacking the enzyme; therefore, glycogen accumulates, eventually in very large amounts. In defects of the debrancher, the breakdown of glycogen stops when 5 glucose units still are present near the branch point in any direction. Short branches of glycogen accumulate.
Glycolysis. The names of sugars are given with a "-" to separate the phosphate moiety so as to emphasize the name of the sugar at each step. As the first step in glycolysis, glucose must enter the cell. Entry is highly dependent on insulin and the reactions at and adjacent to the cell membrane, which are induced by insulin. Next, glucose is given a phosphate handle, so the enzymes of glycolysis (and those of the pentose shunt, glycogen synthesis, further metabolic steps) can handle glucose. When glucose comes from glycogen, it is in the form of glucose-1-phosphate, which has to be converted to glucose-6-phosphate. The glucose moiety is isomerized to fructose, and a second phosphate handle is added to the 6-carbon chain. The chain is then split into two 3-carbon units, each with a phosphate handle. Only one of the 2 forms of P-3-carbon can be metabolized further. The inert form, dihydroxyacetone phosphate, is converted into the active form via the enzyme triosephosphate isomerase. Two molecules of ATP are used up in giving the 6-carbon chain its 2 handles. Nicotine adenine diphosphate (NAD+) is reduced to NADH in the dehydrogenase reaction. In pure glycolysis, these are restored in the oxidative reactions that convert the triose phosphates to pyruvate and the reduction of pyruvate to lactic acid. At the same time, since 2 3-carbon moieties are oxidized for each single 6-carbon moiety that goes down the pathway, 2 extra ATPs are formed. Pure glycolysis thus can generate energy anaerobically. In aerobic metabolism, the bulk of the pyruvate is converted to acetyl coenzyme A rather than to lactate.
Deficiency of the glycogen debrancher enzyme, amylo-1,6-glucosidase, can be demonstrated by direct assay of biopsies from muscle or liver.
Defects of 3 of the 4 proteins that make up the enzyme phosphorylase kinase can be demonstrated directly in liver. In the X-linked form, muscle enzyme is normal, but the enzyme in liver, RBCs, WBCs, and skin fibroblasts is deficient. In other variants, both liver and muscle enzymes are deficient; in yet another, only the muscle enzyme is deficient.
Deficiency of glucose-6-phosphatase (ie, von Gierke disease) has been discussed above in Episodic lactic acidosis in infancy, failure to thrive, and hypotonia with or without features suggesting specific disorders.
A defect of acid maltase or alpha-glucosidase causes Pompe disease, the lysosomal disorder of glycogen. This can be assayed in WBCs, cultured fibroblasts (including those from amniotic fluid), or muscle tissue biopsies.
Treatment
The problems of hypoglycemia due to defects of glycogen's breakdown can be mitigated by giving a nearly constant supply of glucose. In the case of glucose-6-phosphatase defects, this may mean nasogastric administration of glucose or starch solution during the night or, in children who can swallow semisolids, uncooked cornstarch meals. The latter slowly releases glucose when in the gut. Other complications of these diseases, especially of the defects of glucose-6-phosphatase, require more sophisticated measures. Clinical evidence suggests that affected individuals are prone to recurrent episodes of epistaxis and prolonged bleeding times due to poor platelet aggregation secondary to hypoglycemia in the absence of thrombocytopenia or liver dysfunction. Therefore, patients scheduled for surgery need to be monitored closely for bleeding, and consultation with a metabolic and/or hematologic expert is advisable prior to elective procedures in these children.
Episodic vomiting in infants or young children
While episodic vomiting has many causes, disorders of fructose metabolism should be kept in mind. These almost invariably are associated with hypoglycemia, because errors of fructose metabolism interfere with glycogenolysis (ie, breakdown of glycogen to glucose-6-phosphate and then to pyruvate) and with gluconeogenesis (ie, breakdown of pyruvate to glucose-6-phosphate). Children whose hereditary fructose intolerance is mild enough to allow them to survive infancy generally vomit shortly after eating fructose (ie, any source of sucrose or fruit). Many of them learn to avoid these foods and abhor candy. Children with a specific defect of fructose 1-phosphate aldolase may have a similar aversion (see the image below).
Fructose metabolism. In muscle and most tissue, the same hexokinase that phosphorylates glucose to glucose-6-phosphate also phosphorylates fructose to fructose-6-phosphate. Fructose-6- phosphate then undergoes metabolism, down to pyruvate or up to glycogen, through the enzymes of glycolysis. In the liver, fructose is phosphorylated largely to the 1-phosphate form and converted to glyceraldehydes and dihydroxyacetone phosphate. Free glyceraldehydes can be phosphorylated by a specific kinase; this and dihydroxyacetone are metabolized further by the reactions of glycolysis.
Ingesting something that induces vomiting is a powerful negative psychological stimulus in all mammals (hence the aversion to sweet alcoholic drinks in adults whose first ventures with alcohol led to vomiting, when they first drank sweet liquors in high school or college).
Differential diagnosis
Episodic vomiting in infants or young children has a large number of causes, most of which are more common than disorders of carbohydrate metabolism. Nonmetabolic causes include gastroenteritis from viruses, bacteria, and parasites; gastroesophageal reflux disorder; irritable bowel syndrome; small or large bowel obstructions; neurological conditions, such as premigrainous syndromes and brain masses; and abdominal epilepsy. In this clinical setting, inborn errors of the urea cycle and organic acid metabolism need to be considered first, before considering lactose intolerance from inherited disorders of galactose and fructose metabolism.
Workup and treatment
Hereditary fructose intolerance and defects of fructose 1, 6-diphosphate aldolase are discussed under Episodic lactic acidosis in infancy, failure to thrive, and hypotonia with or without features suggesting specific disorders earlier in this article. Disorders of galactose metabolism are discussed in the subsection Infants with intellectual disability, hypotonia, failure to thrive, and other features.
Episodic acidosis in childhood or early adult life
This often presents with features of a specific disorder, including many of the differential diagnoses mentioned under Episodic lactic acidosis in infancy, failure to thrive, and hypotonia with or without features suggesting specific disorders. Specific features may enable rapid diagnosis.
Intermittent or episodic ataxia in childhood and adolescence
This category is characterized by the sudden, almost abrupt, onset of ataxia, extrapyramidal symptoms in the upper extremities, and dysarthria soon after or in conjunction with an infectious illness or severe emotional upset. In one patient monitored by one of the authors, death of a grandparent and critical school examinations were sufficient stimuli to initiate attacks of ataxia. Physical findings in this patient also included a high arched palate, minimal kyphoscoliosis, minimal pes cavus, and extra rows of lashes on the upper eyelid. When the boy was 8 or 10 years old, a mild attack was precipitated by an environmental challenge with flashing red lights from scintillation counters and Geiger counters for about 30 minutes in a dimly lit room. For this patient and others, the episodes of ataxia usually last about 2 weeks. Although the neurologic symptoms usually are accompanied by increased serum lactic acid, frank acidosis is not always present.
The underlying defect in this patient was shown to be that of the first enzyme of the PDH complex, E1. Eventually, he was placed on a diet high in thiamine and relatively low in carbohydrates; the number of attacks subsided. While this patient graduated from college, others with similar clinical presentations have had at least a mild degree of intellectual disability.
Work in the 1970s and early 1980s raised the question whether familial cases of a childhood- or adolescent-onset ataxia (one with slower progression than is seen in classical Friedreich ataxia) was associated with moderate defects of the activity of PDH, E3. The association has not been confirmed. Classic Friedreich ataxia appears to be caused by a single genetic defect and does not appear to be associated with measurable abnormalities in the E3 pathway
Differential diagnosis
Intermittent or episodic ataxia in childhood and adolescence is a rare condition. Porphyria might induce this, and multiple sclerosis, though rare in young people, certainly can present with ataxia; however, multiple sclerosis almost always manifests in other ways. Postinfectious cerebellar dysfunction usually presents as a single episode that clears in weeks or months and does not recur. Griggs and colleagues have described patients with a familial form of intermittent ataxia, unrelated to pyruvate defects, that responds to treatment with acetazolamide.
Workup
E1 is the first enzyme of the PDH multienzyme complex. Phosphorylation inactivates this pyruvate decarboxylase, which activates by removal of the phosphate. The decarboxylase requires thiamine pyrophosphate as a substrate. Patients deficient in this enzyme have elevations of metabolic by-products related to pyruvate degradation during clinical attacks of ataxia (ie, excess lactic acid and alanine in blood or urine, or both). The primary enzyme defect can be demonstrated in cultured fibroblasts and probably can be demonstrated in WBCs or muscle tissue. The substrate, pyruvate, is so labile and the enzyme activity of this and other components of the PDH complex so sensitive to environmental contaminants that carefully controlled assays need to be performed in a research laboratory.
Treatment
The young man discussed earlier in detail was treated with a low-carbohydrate diet and high doses of thiamine. Some patients with autosomal recessive ataxia with large-fiber-posterior column sensory loss, areflexia, cardiomyopathy, and bony stigmata have been treated with some success with a modified (titrated) ketogenic diet. Dietary changes were introduced gradually until moderate to high levels of ketones appeared in the urine without producing frank metabolic acidosis.
Cramps and weakness, often with some episodes of muscle breakdown and myoglobinuria, during or after heavy exercise
Black McArdle first described these symptoms of the disease at Guy's Hospital in London. [16] (He was so named to distinguish him from his brother "Red," also a distinguished physician there; the pseudonyms referred to the color of their hair.) Type V glycogenosis due to muscle phosphorylase deficiency is the clinically mildest of the glycogen storage diseases, and the symptoms may not occur until adolescence or young adulthood.
The symptoms tend to be induced during exercise when muscle depends on glycolysis rather than oxidative phosphorylation as a source of fuel. Exercise that includes sprinting or vigorous running most likely will induce symptoms. Often, onset of symptoms comes during what normally would represent the subjective "second wind." Over time, patients may learn to slow down or stop at the first hint of symptoms, and then to begin again, with what has been called a second wind but which is probably a pause in which oxidative metabolism can generate sufficient ATP in muscle so that anaerobic activity can begin again.
If this speculation is true, the "second wind" that is peculiar to myopathies from glycolytic/glycogenolytic defects differs in its nature from the second wind experienced by healthy athletes. The patients therefore can carry on anaerobic exercise in spurts over a long time, presumably with aerobic exercise in between. Affected individuals often can tailor their activities to take a break at the first sign of cramping, and then resume activities shortly thereafter.
Without this pause and resumption in athletes with glycolytic/glycogenolytic defects, muscles suddenly become so weak that the athlete becomes virtually immobile from painful cramps, and the muscles that were exercised are very sore and tender. This disease and the finding of elevated lactic acid levels in the blood after exercise led to the erroneous notion that muscle cramps in general are caused by lactic acidosis. The muscles in the individuals affected with glycolytic defects continue to be very sore even after the cramps resolve.
If the urine is not frankly red, it must be examined to see if it still contains heme groups. In myopathy due to disorders of glycolysis, the heme pigment proves to be myoglobin, not hemoglobin, when detailed analyses are carried out. With McArdle disease, muscle enzymes and creatine levels usually are elevated in the blood after a symptomatic episode. In this condition, muscle histology demonstrates marked increase in glycogen stores in the myofibrils. Histochemical or immunoassays for specific enzymes can augment laboratory diagnosis by identifying the precise enzyme defect.
The original enzyme defect described by McArdle [16] and then by Schmid and Mahler [17] was localized to the glycolysis pathway. The deficiency ultimately was identified in muscle phosphorylase. Through the past decade, MR spectroscopy of forearm muscles in vivo has led to the diagnosis. Deficiencies of several other glycolytic enzymes (eg, muscle phosphofructokinase, phosphoglycerate kinase, phosphoglycerate mutase, lactate dehydrogenase) produce similar clinical pictures and are indistinguishable from muscle phosphorylase deficiency, unless enzymatic tests are performed.
Muscle breakdown occurs in disorders of glycolysis and glycogenolysis during muscle activity that is largely dependent on glycolysis or anaerobic metabolism. This is in striking contrast to the circumstances of muscle breakdown in disorders related to lipid or ketone metabolism. In these disorders, muscle breaks down, with similar symptoms, under circumstances in which muscles are relying on oxidative metabolism, especially of ketone bodies, as a fuel source. Activity most likely to produce symptoms under these circumstances is prolonged, less vigorous exercise over several hours, which is exacerbated by starvation or high-fat, high-protein meals and occurs more often in a cold or cool environment. Histologically, myofibrils may demonstrate increased lipid droplets, but in contrast to the glycogenoses, glycogen stores are either normal or even slightly depleted.
Differential diagnosis
The differential diagnosis includes all the acute and symptomatic myopathies, including various forms of polymyositis. Many of these appear to be viral or postinfectious; still others appear to be paraneoplastic or associated with motor neuron degenerations or systemic collagen-vascular diseases but not with problems of intermediary metabolism.
The circumstances under which cramps and weakness occur may provide insight into whether a metabolic disorder is present. If evidence of metabolic etiology appears, differentiating between disorders affecting white muscle (ie, the carbohydrate errors discussed here) and disorders affecting red muscle (ie, errors of lipid and mitochondrial metabolism) is essential, because these disorders can present similar pictures but require very different metabolic treatment strategies. Making the correct diagnosis clearly is important since, for each of these groups of disorders of glycolysis/glycogenolysis, mitochondrial transport, and oxidative phosphorylation, treatment approaches are quite different.
Workup
Prior to the availability of direct enzyme assays, defects of muscle phosphorylase had to be inferred from the clinical features of muscle cramping with vigorous exercise in conjunction with accumulation of lactic acid in the blood and myoglobin in the urine. The forearm ischemia test further substantiated the diagnosis. For this test, a blood pressure cuff is inflated to systolic pressure. With the cuff on, ampules of venous blood are drawn from the antecubital fossa via an indwelling catheter attached to a stopcock before, during, and after 1 minute of forearm exercise (eg, having the patient squeeze a partially inflated blood-pressure cuff repeatedly). In reality, many patients with phosphorylase deficiency cannot continue to exercise for a full minute. Blood samples are assayed for glucose, lactic acid, and sometimes pyruvate. Affected individuals have lower glucose levels and higher lactate and pyruvate levels compared to reference values in blood samples taken 1-2 minutes after exercise.
In the last 25 years, the preferred diagnostic test for McArdle disease has been a phosphorylase assay directly on muscle tissue. Today, histochemical or biochemical techniques can be used to diagnose deficiencies in even the rare enzymes that clinically simulate McArdle disease. Again, these enzymes include muscle phosphofructokinase, phosphoglycerate kinase, phosphoglycerate mutase, and lactate dehydrogenase. In the past 2 decades, diagnosis also has been made by analysis of metabolites in vivo on human forearm muscle by means of MR spectroscopy.
Treatment
DiMauro et al gave a thorough review of strategies to treat disorders of muscle phosphorylase. [18] Pyridoxine, at 50 mg/day, seems to be effective. (Note that at 100 mg/d, this vitamin can produce a painful distal sensory neuropathy.) High-protein diets also were effective, at least for several weeks, but determining the long-term success of dietary treatment will be possible only after adequate clinical trials.
Current therapeutic options include a carbohydrate-rich diet and carbohydrate ingestion shortly before strenuous exercise in combination with medically supervised aerobic training of low to moderate intensity.
Symmetrical neuromuscular disease with weakness and wasting of proximal muscles
Only rarely are cases of progressive neuromuscular diseases caused by defects of glycogen metabolism. When they are, children or young adults generally develop, over months or years, progressive weakness of the limb-girdle muscles and the proximal muscles of the limbs. Patients have no pain under ordinary circumstances, but heavy exercise may induce fatigue and cramps. Symptoms depend on the nature of the underlying metabolic defect. Muscles may or may not be atrophied at presentation but eventually waste. Patients do not complain of muscle tenderness unless cramping occurs. Some patients with debrancher deficiency have evidence of cirrhosis; others become asymptomatic in adult life.
Differential diagnosis
This condition is essentially indistinguishable from Limb-Girdle Muscular Dystrophy. The symptoms can be demonstrated in a number of acquired diseases, including various forms of polymyositis or inflammatory myopathy, various neuropathic diseases (especially proximal forms of motor neuron disease and the limb-girdle pattern of painless diabetic multiple neuropathy or diabetic plexitis), inherited mitochondrial myopathies (often associated with ragged red fibers), disorders of lipid metabolism, and/or defects of lipid as carnitine transport at the level of mitochondria. For lipids to enter the mitochondria, they need to be complexed with carnitine derivatives. Primary or secondary carnitine deficiencies as well as deficiencies of enzymes involved in the metabolic pathway can interfere with this process, resulting in a myopathic picture.
Workup
Muscle enzyme levels may be elevated in the serum, but myoglobinuria is an unusual finding. Histochemical analysis of biopsied muscle shows excessive glycogen stores. Partial acid maltase deficiency may result in the adult form of Pompe disease and may be clinically indistinguishable from muscle phosphofructokinase deficiency (reported in at least one case) and glycogen debrancher enzyme deficiency. However, patients with glycogen debrancher enzyme deficiency eventually develop physical, electrical, or radiographic evidence of cardiac ventricle dilatation. Therefore, clinicians are advised to monitor for these changes by ECGs and examinations on an annual basis.
Treatment
The FDA designated alglucosidase alfa (Myozyme) an orphan drug for use in infants with Pompe disease.
The long-term health impact of consumption of nonnutritive sweeteners is uncertain. Azad et al. (2017) conclude that evidence from RCTs offer no clear support for the intended benefits of nonnutritive sweeteners for weight management. They further add that observational data suggest that routine intake of nonnutritive sweeteners may be associated with increased BMI and cardiometabolic risk. Further research is needed to fully characterize the long-term risks and benefits of nonnutritive sweeteners. [19]
Williamson (2017) discusses the role of polyphenols in modern nutrition. [20] Polyphenols are found in plant‐based foods and beverages, notably apples, berries, citrus fruit, plums, broccoli, cocoa, tea and coffee and many others. There is enough epidemiological evidence that a diet high in polyphenol‐rich fruit, vegetables, cocoa, and beverages offer protection against developing cardiovascular disease and type 2 diabetes. Common polyphenols in the diet are flavanols (cocoa, tea, apples, broad beans), flavanones (hesperidin in citrus fruit), hydroxycinnamates (coffee, many fruits), flavonols (quercetin in onions, apples and tea), and anthocyanins (berries). Coffee and tea both reduce the risk of developing type 2 diabetes through action of their constituent polyphenols by an unknown mechanism. Obviously more research is needed in this area to make strong recommendations.
A database maintained by King Abdullah University of Science and Technology and University of Cambridge exists called Drug Database for Inborn Errors of Metabolism (DDIEM).
Future in Treatment
In many instances, inborn errors of metabolism are difficult or impossible to treat. Novel therapeutic modalities, including gene therapy, need to be explored. Among available gene delivery systems, recombinant adeno-associated virus (AAV) and CRISPR/Cas9 genome editing shows promise for the treatment of metabolic disease. Despite the relatively low immunogenicity of AAV vectors, immune responses are also emerging as a factor requiring special attention as efforts accelerate toward human clinical translation. Trials targeting lipoprotein lipase deficiency are showing early evidence of efficacy. [21]
Fertility Concerns
Increasing numbers of individuals with inherited metabolic disorders are surviving into adulthood and considering their reproductive options. Lee discusses a practical approach to supporting such individuals, focusing on issues concerning fertility, the impact of pregnancy on metabolism and the metabolic disorder itself on the pregnancy, as well as highlighting the need to pay special attention during the postpartum period. [22] He further opines that apart from pregnancies in women with phenylketonuria, data are lacking in this area. Collection of information within registries is needed to aid our understanding of potential problems and counseling of women and their partners.
-
Ketone metabolism.
-
Krebs cycle.
-
Metabolism of galactose. The galactose ring differs from glucose in that the hydroxyl group (OH) at carbon-3 is up and not down. To convert galactose to glucose, the ring is first bound to uridine diphosphate (UDP). In this form, the sugar moiety can be epimerized, putting the OH down and forming glucose. The UDP-glucose then is converted to glucose-1-phosphate, then to glucose-6-phosphate, and subsequently metabolized further.
-
Structure of glycogen. The rings each indicate a glucose moiety in glycogen. These are linked by an ether bond between carbon-1 of one moiety and carbon-4 of the next. In the hydrolysis of glycogen, glycogen phosphorylase (ie, phosphorylase) catalyses the hydrolysis of the glucose moiety from the nonreducing end of glycogen to glucose-1-phosphate and a glycogen chain 1 unit shorter in length than the original. Phosphoglucomutase converts the glucose-1-phosphate to glucose-6-phosphate. Glucose-1-phosphate can then be metabolized, eg, by glycolysis. The branches of glycogen comprise 1-6 ether links. These have to be broken by the glycogen debranching enzyme. In fact, the debranching enzyme has to act before phosphorylase can hydrolyze moieties within 5 units of a branch point. In defects of phosphorylase, glycogen cannot be broken down in the tissue lacking the enzyme; therefore, glycogen accumulates, eventually in very large amounts. In defects of the debrancher, the breakdown of glycogen stops when 5 glucose units still are present near the branch point in any direction. Short branches of glycogen accumulate.
-
Glycolysis. The names of sugars are given with a "-" to separate the phosphate moiety so as to emphasize the name of the sugar at each step. As the first step in glycolysis, glucose must enter the cell. Entry is highly dependent on insulin and the reactions at and adjacent to the cell membrane, which are induced by insulin. Next, glucose is given a phosphate handle, so the enzymes of glycolysis (and those of the pentose shunt, glycogen synthesis, further metabolic steps) can handle glucose. When glucose comes from glycogen, it is in the form of glucose-1-phosphate, which has to be converted to glucose-6-phosphate. The glucose moiety is isomerized to fructose, and a second phosphate handle is added to the 6-carbon chain. The chain is then split into two 3-carbon units, each with a phosphate handle. Only one of the 2 forms of P-3-carbon can be metabolized further. The inert form, dihydroxyacetone phosphate, is converted into the active form via the enzyme triosephosphate isomerase. Two molecules of ATP are used up in giving the 6-carbon chain its 2 handles. Nicotine adenine diphosphate (NAD+) is reduced to NADH in the dehydrogenase reaction. In pure glycolysis, these are restored in the oxidative reactions that convert the triose phosphates to pyruvate and the reduction of pyruvate to lactic acid. At the same time, since 2 3-carbon moieties are oxidized for each single 6-carbon moiety that goes down the pathway, 2 extra ATPs are formed. Pure glycolysis thus can generate energy anaerobically. In aerobic metabolism, the bulk of the pyruvate is converted to acetyl coenzyme A rather than to lactate.
-
Scheme of the major reactions of the pyruvate dehydrogenase complex. Initial oxidative decarboxylation of pyruvate is shown as reaction (1), transfer of the 2-carbon moiety to the lipoyl side chain of lipoyl acetyltransferase (E2) as reaction (2), formation of acetyl coenzyme A as reaction (3), oxidation of the lipoyl moiety as reaction (4), and reduction of NAD as reaction (5). The regulatory enzymes acting on pyruvate decarboxylase (E1) are not shown.
-
Major metabolic relations of pyruvate. Many of the reactions, such as those between glucose-6-phosphate and pyruvate, pyruvate and alanine, and pyruvate and lactate, are of course reversible. Abbreviations: LDH - lactic dehydrogenase; PDH - pyruvate dehydrogenase complex; p carboxylase - pyruvate carboxylase; TCA cycle - Krebs tricarboxylic acid cycle; KGDH-a-ketoglutarate dehydrogenase complex.
-
Fructose metabolism. In muscle and most tissue, the same hexokinase that phosphorylates glucose to glucose-6-phosphate also phosphorylates fructose to fructose-6-phosphate. Fructose-6- phosphate then undergoes metabolism, down to pyruvate or up to glycogen, through the enzymes of glycolysis. In the liver, fructose is phosphorylated largely to the 1-phosphate form and converted to glyceraldehydes and dihydroxyacetone phosphate. Free glyceraldehydes can be phosphorylated by a specific kinase; this and dihydroxyacetone are metabolized further by the reactions of glycolysis.







