Sadowski M, Verma A, Wisniewski T. Prion Diseases. Bradley WG, Daroff RB, Fenichel GM, Jankovic J, eds. Neurology in Clinial Practice. Philadelphia: Elsevier Inc; 2004. 1613-1630.
Takada LT, Geschwind MD. Prion diseases. Semin Neurol. 2013 Sep. 33(4):348-56. [QxMD MEDLINE Link].
Gordon WS. Advances in veterinary research. Vet Rec. 1946. 58:518-525.
Griffith JS, Hadlow WJ. Scrapie and kuru. Lancet. 1959. 11:289-290.
Prusiner SB, Scott MR, DeArmond SJ, Cohen FE. Prion protein biology. Cell. 1998 May 1. 93(3):337-48. [QxMD MEDLINE Link].
Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science. 1982 Apr 9. 216(4542):136-44. [QxMD MEDLINE Link].
Vanni I, Pirisinu L, Acevedo-Morantes C, Kamali-Jamil R, Rathod V, Di Bari MA, et al. Isolation of infectious, non-fibrillar and oligomeric prions from a genetic prion disease. Brain. 2020 May 1. 143 (5):1512-1524. [QxMD MEDLINE Link].
Weissmann C. The Ninth Datta Lecture. Molecular biology of transmissible spongiform encephalopathies. FEBS Lett. 1996 Jun 24. 389(1):3-11. [QxMD MEDLINE Link].
Lathe R, Darlix JL. Prion protein PrP nucleic acid binding and mobilization implicates retroelements as the replicative component of transmissible spongiform encephalopathy. Arch Virol. 2020 Mar. 165 (3):535-556. [QxMD MEDLINE Link].
Clavaguera F, Bolmont T, Crowther RA, Abramowski D, Frank S, Probst A. Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol. 2009 Jul. 11(7):909-13. [QxMD MEDLINE Link]. [Full Text].
Frost B, Jacks RL, Diamond MI. Propagation of tau misfolding from the outside to the inside of a cell. J Biol Chem. 2009 May 8. 284(19):12845-52. [QxMD MEDLINE Link]. [Full Text].
Ren PH, Lauckner JE, Kachirskaia I, Heuser JE, Melki R, Kopito RR. Cytoplasmic penetration and persistent infection of mammalian cells by polyglutamine aggregates. Nat Cell Biol. 2009 Feb. 11(2):219-25. [QxMD MEDLINE Link]. [Full Text].
Thackray AM, Knight R, Haswell SJ, Bujdoso R, Brown DR. Metal imbalance and compromised antioxidant function are early changes in prion disease. Biochem J. 2002 Feb 15. 362:253-8. [QxMD MEDLINE Link].
Harris DA, Lele P, Snider WD. Localization of the mRNA for a chicken prion protein by in situ hybridization. Proc Natl Acad Sci U S A. 1993 May 1. 90(9):4309-13. [QxMD MEDLINE Link]. [Full Text].
Windl O, Dempster M, Estibeiro P, Lathe R. A candidate marsupial PrP gene reveals two domains conserved in mammalian PrP proteins. Gene. 1995 Jul 4. 159(2):181-6. [QxMD MEDLINE Link].
Masison DC, Edskes HK, Maddelein ML, Taylor KL, Wickner RB. [URE3] and [PSI] are prions of yeast and evidence for new fungal prions. Curr Issues Mol Biol. 2000 Apr. 2(2):51-9. [QxMD MEDLINE Link].
Büeler H, Aguzzi A, Sailer A, et al. Mice devoid of PrP are resistant to scrapie. Cell. 1993 Jul 2. 73(7):1339-47. [QxMD MEDLINE Link].
Collinge J, Whittington MA, Sidle KC, et al. Prion protein is necessary for normal synaptic function. Nature. 1994 Jul 28. 370(6487):295-7. [QxMD MEDLINE Link].
Tobler I, Gaus SE, Deboer T, et al. Altered circadian activity rhythms and sleep in mice devoid of prion protein. Nature. 1996 Apr 18. 380(6575):639-42. [QxMD MEDLINE Link].
Riek R, Hornemann S, Wider G, Billeter M, Glockshuber R, Wuthrich K. NMR structure of the mouse prion protein domain PrP(121-321). Nature. 1996 Jul 11. 382(6587):180-2. [QxMD MEDLINE Link].
Hosszu LL, Baxter NJ, Jackson GS, et al. Structural mobility of the human prion protein probed by backbone hydrogen exchange. Nat Struct Biol. 1999 Aug. 6(8):740-3. [QxMD MEDLINE Link].
James TL, Liu H, Ulyanov NB, et al. Solution structure of a 142-residue recombinant prion protein corresponding to the infectious fragment of the scrapie isoform. Proc Natl Acad Sci U S A. 1997 Sep 16. 94(19):10086-91. [QxMD MEDLINE Link]. [Full Text].
Knaus KJ, Morillas M, Swietnicki W, Malone M, Surewicz WK, Yee VC. Crystal structure of the human prion protein reveals a mechanism for oligomerization. Nat Struct Biol. 2001 Sep. 8(9):770-4. [QxMD MEDLINE Link].
Aucouturier P, Geissmann F, Damotte D, et al. Infected splenic dendritic cells are sufficient for prion transmission to the CNS in mouse scrapie. J Clin Invest. 2001 Sep. 108(5):703-8. [QxMD MEDLINE Link].
Pan KM, Baldwin M, Nguyen J, et al. Conversion of alpha-helices into beta-sheets features in the formation of the scrapie prion proteins. Proc Natl Acad Sci U S A. 1993 Dec 1. 90(23):10962-6. [QxMD MEDLINE Link]. [Full Text].
Baron T. Identification of inter-species transmission of prion strains. J Neuropathol Exp Neurol. 2002 May. 61(5):377-83. [QxMD MEDLINE Link].
Kascsak RJ, Rubenstein R, Merz PA, et al. Immunological comparison of scrapie-associated fibrils isolated from animals infected with four different scrapie strains. J Virol. 1986 Sep. 59(3):676-83. [QxMD MEDLINE Link]. [Full Text].
Carp RI, Rubenstein R. Diversity and significance of scrapie strains. Semin Virol. 1991. 2:203-13.
Bessen RA, Marsh RF. Distinct PrP properties suggest the molecular basis of strain variation in transmissible mink encephalopathy. J Virol. 1994 Dec. 68(12):7859-68. [QxMD MEDLINE Link]. [Full Text].
Caughey B, Raymond GJ, Bessen RA. Strain-dependent differences in beta-sheet conformations of abnormal prion protein. J Biol Chem. 1998 Nov 27. 273(48):32230-5. [QxMD MEDLINE Link].
Parchi P, Castellani R, Capellari S, et al. Molecular basis of phenotypic variability in sporadic Creutzfeldt-Jakob disease. Ann Neurol. 1996 Jun. 39(6):767-78. [QxMD MEDLINE Link].
Collinge J, Sidle KC, Meads J, Ironside J, Hill AF. Molecular analysis of prion strain variation and the aetiology of 'new variant' CJD. Nature. 1996 Oct 24. 383(6602):685-90. [QxMD MEDLINE Link].
Kimberlin RH, Walker CA. Evidence that the transmission of one source of scrapie agent to hamsters involves separation of agent strains from a mixture. J Gen Virol. 1978 Jun. 39(3):487-96. [QxMD MEDLINE Link].
Nicoll AJ, Collinge J. Preventing prion pathogenicity by targeting the cellular prion protein. Infect Disord Drug Targets. 2009 Feb. 9(1):48-57. [QxMD MEDLINE Link].
Aucouturier P, Carp RI, Carnaud C, Wisniewski T. Prion diseases and the immune system. Clin Immunol. 2000 Aug. 96(2):79-85. [QxMD MEDLINE Link].
Eklund CM, Kennedy RC, Hadlow WJ. Pathogenesis of scrapie virus infection in the mouse. J Infect Dis. 1967 Feb. 117(1):15-22. [QxMD MEDLINE Link].
Fraser H, Dickinson AG. Studies of the lymphoreticular system in the pathogenesis of scrapie: the role of spleen and thymus. J Comp Pathol. 1978 Oct. 88(4):563-73. [QxMD MEDLINE Link].
Kimberlin RH, Walker CA. Pathogenesis of mouse scrapie: dynamics of agent replication in spleen, spinal cord and brain after infection by different routes. J Comp Pathol. 1979 Oct. 89(4):551-62. [QxMD MEDLINE Link].
Houston F, Foster JD, Chong A, Hunter N, Bostock CJ. Transmission of BSE by blood transfusion in sheep. Lancet. 2000 Sep 16. 356(9234):999-1000. [QxMD MEDLINE Link].
Klein MA, Frigg R, Flechsig E, et al. A crucial role for B cells in neuroinvasive scrapie. Nature. 1997 Dec 18-25. 390(6661):687-90. [QxMD MEDLINE Link].
Montrasio F, Frigg R, Glatzel M, et al. Impaired prion replication in spleens of mice lacking functional follicular dendritic cells. Science. 2000 May 19. 288(5469):1257-9. [QxMD MEDLINE Link].
Shlomchik MJ, Radebold K, Duclos N, Manuelidis L. Neuroinvasion by a Creutzfeldt-Jakob disease agent in the absence of B cells and follicular dendritic cells. Proc Natl Acad Sci U S A. 2001 Jul 31. 98(16):9289-94. [QxMD MEDLINE Link]. [Full Text].
Harischandra DS, Kondru N, Martin DP, Kanthasamy A, Jin H, Anantharam V, et al. Role of proteolytic activation of protein kinase Cd in the pathogenesis of prion disease. Prion. 2014 Jan-Feb. 8(1):143-53. [QxMD MEDLINE Link].
Beekes M, McBride PA, Baldauf E. Cerebral targeting indicates vagal spread of infection in hamsters fed with scrapie. J Gen Virol. 1998 Mar. 79 ( Pt 3):601-7. [QxMD MEDLINE Link].
Glatzel M, Heppner FL, Albers KM, Aguzzi A. Sympathetic innervation of lymphoreticular organs is rate limiting for prion neuroinvasion. Neuron. 2001 Jul 19. 31(1):25-34. [QxMD MEDLINE Link].
Maddox RA, Person MK, Blevins JE, Abrams JY, Appleby BS, Schonberger LB, et al. Prion disease incidence in the United States: 2003-2015. Neurology. 2020 Jan 14. 94 (2):e153-e157. [QxMD MEDLINE Link].
Uttley L, Carroll C, Wong R, Hilton DA, Stevenson M. Creutzfeldt-Jakob disease: a systematic review of global incidence, prevalence, infectivity, and incubation. Lancet Infect Dis. 2020 Jan. 20 (1):e2-e10. [QxMD MEDLINE Link].
Meiner Z, Halimi M, Polakiewicz RD, Prusiner SB, Gabizon R. Presence of prion protein in peripheral tissues of Libyan Jews with Creutzfeldt-Jakob disease. Neurology. 1992 Jul. 42(7):1355-60. [QxMD MEDLINE Link].
Neufeld MY, Josiphov J, Korczyn AD. Demyelinating peripheral neuropathy in Creutzfeldt-Jakob disease. Muscle Nerve. 1992 Nov. 15(11):1234-9. [QxMD MEDLINE Link].
Holman RC, Khan AS, Belay ED, Schonberger LB. Creutzfeldt-Jakob disease in the United States, 1979-1994: using national mortality data to assess the possible occurrence of variant cases. Emerg Infect Dis. 1996 Oct-Dec. 2(4):333-7. [QxMD MEDLINE Link]. [Full Text].
Masters CL, Harris JO, Gajdusek DC, Gibbs CJ Jr, Bernoulli C, Asher DM. Creutzfeldt-Jakob disease: patterns of worldwide occurrence and the significance of familial and sporadic clustering. Ann Neurol. 1979 Feb. 5(2):177-88. [QxMD MEDLINE Link].
Cathala F, Baron H. Clinical Aspects of Creutzfeldt-Jakob Disease. Prusiner SB, McKinley MP, eds. Prions: novel infectious pathogens causing scrapie and Creutzfeldt-Jakob disease. New York, NY: Academic Press; 1987. 467-509.
Gajdusek DC, Zigas V. Degenerative disease of the central nervous system in New Guinea: the epidemic occurrence of "kuru" in the native population. N Engl J Med. 1957. 257:974-978.
Gajdusek DC, Zigas V. Clinical, pathological and epidemiological study of an acute progressive degenerative disease of the central nervous system among natives of the eastern highlands of New Guinea. Am J Med. 1959. 26:442-469.
Liberski PP, Sikorska B, Lindenbaum S, Goldfarb LG, McLean C, Hainfellner JA, et al. Kuru: Genes, Cannibals and Neuropathology. J Neuropathol Exp Neurol. 2012 Feb. 71(2):92-103. [QxMD MEDLINE Link].
Gajdusek DC, Gibbs CJ, Alpers M. Experimental transmission of a Kuru-like syndrome to chimpanzees. Nature. 1966 Feb 19. 209(5025):794-6. [QxMD MEDLINE Link].
Jacob A. Uber eigenaritge erkrankungen des zentral-nervensystems mit bemerkenswertem anatomischen befunde (spastische pseudosklerose-encephalomyelopathie mit disseminierten degenerationsherden). Z Gesamte Neurol Psychiatre. 1921. 64:147-228.
Hofmann J, Wolf H, Grassmann A, Arndt V, Graham J, Vorberg I. Creutzfeldt-Jakob disease and mad cows: lessons learnt from yeast cells. Swiss Med Wkly. 2012 Jan 24. 142:[QxMD MEDLINE Link].
Baiardi S, Redaelli V, Ripellino P, Rossi M, Franceschini A, Moggio M, et al. Prion-related peripheral neuropathy in sporadic Creutzfeldt-Jakob disease. J Neurol Neurosurg Psychiatry. 2019 Apr. 90 (4):424-427. [QxMD MEDLINE Link].
Chiofalo N, Fuentes A, Galvez S. Serial EEG findings in 27 cases of Creutzfeldt-Jakob disease. Arch Neurol. 1980 Mar. 37(3):143-5. [QxMD MEDLINE Link].
Gerstmann J, Straussler E, Scheinker I. Über eine eigenartige hereditar-familiare Erkrankung des Zentralnervensystems zugleich ein Beitrag zür frage des vorzeitigen kokalen Alterns. Z Neurol. 1936. 154:736-762.
Ghetti B, Tagliavini F, Giaccone G, et al. Familial Gerstmann-Sträussler-Scheinker disease with neurofibrillary tangles. Mol Neurobiol. 1994 Feb. 8(1):41-8. [QxMD MEDLINE Link].
Ghetti B, Piccardo P, Spillantini MG, et al. Vascular variant of prion protein cerebral amyloidosis with tau-positive neurofibrillary tangles: the phenotype of the stop codon 145 mutation in PRNP. Proc Natl Acad Sci U S A. 1996 Jan 23. 93(2):744-8. [QxMD MEDLINE Link]. [Full Text].
Medori R, Tritschler HJ, LeBlanc A, et al. Fatal familial insomnia, a prion disease with a mutation at codon 178 of the prion protein gene. N Engl J Med. 1992 Feb 13. 326(7):444-9. [QxMD MEDLINE Link].
Mastrangelo V, Merli E, Rucker JC, Eggenberger ER, Zee DS, Cortelli P. Neuro-Ophthalmological Findings in Early Fatal Familial Insomnia. Ann Neurol. 2021 Apr. 89 (4):823-827. [QxMD MEDLINE Link].
Goldfarb LG, Brown P, Haltia M, et al. Creutzfeldt-Jakob disease cosegregates with the codon 178Asn PRNP mutation in families of European origin. Ann Neurol. 1992 Mar. 31(3):274-81. [QxMD MEDLINE Link].
Solvason HB, Harris B, Zeifert P, Flores BH, Hayward C. Psychological versus biological clinical interpretation: a patient with prion disease. Am J Psychiatry. 2002 Apr. 159(4):528-37. [QxMD MEDLINE Link].
Oliveros RG, Saracibar N, Gutierrez M, , Munon T, Gonzalez-Pinto A. Catatonia due to a prion familial disease. Schizophr Res. 2009 Mar. 108(1-3):309-10. [QxMD MEDLINE Link].
Collinge J. Human prion diseases and bovine spongiform encephalopathy (BSE). Hum Mol Genet. 1997. 6(10):1699-705. [QxMD MEDLINE Link].
Collinge J, Rossor M. A new variant of prion disease. Lancet. 1996 Apr 6. 347(9006):916-7. [QxMD MEDLINE Link].
Bateman D, Hilton D, Love S, Zeidler M, Beck J, Collinge J. Sporadic Creutzfeldt-Jakob disease in a 18-year-old in the UK. Lancet. 1995 Oct 28. 346(8983):1155-6. [QxMD MEDLINE Link].
Britton TC, al-Sarraj S, Shaw C, Campbell T, Collinge J. Sporadic Creutzfeldt-Jakob disease in a 16-year-old in the UK. Lancet. 1995 Oct 28. 346(8983):1155. [QxMD MEDLINE Link].
Collee JG, Bradley R. BSE: a decade on--Part I. Lancet. 1997 Mar 1. 349(9052):636-41. [QxMD MEDLINE Link].
Will RG. Surveillance of prion disease in humans. HF Baker, Ridley RM, eds. Methods in Molecular Medicine: Prion Diseases. Totowa, NJ: Humana Press Inc; 1996. 119-137.
Liberski PP, Guiroy DC, Williams ES, Walis A, Budka H. Deposition patterns of disease-associated prion protein in captive mule deer brains with chronic wasting disease. Acta Neuropathol. 2001 Nov. 102(5):496-500. [QxMD MEDLINE Link].
Ciarlariello VB, Barsottini OGP, Espay AJ, Pedroso JL. Arm Levitation as Initial Manifestation of Creutzfeldt-Jakob Disease: Case Report and Review of the Literature. Tremor Other Hyperkinet Mov (N Y). 2018. 8:572. [QxMD MEDLINE Link].
Rodriguez-Porcel F, Ciarlariello VB, Dwivedi AK, Lovera L, Da Prat G, Lopez-Castellanos R, et al. Movement Disorders in Prionopathies: A Systematic Review. Tremor Other Hyperkinet Mov (N Y). 2019. 9:[QxMD MEDLINE Link].
Khan RM, Gunaratne LA, Kinmont JC. Rapid fracture healing in a patient with inherited prion disease. Ann R Coll Surg Engl. 2009 Apr. 91(3):261-2. [QxMD MEDLINE Link].
Peckeu L, Brandel JP, Welaratne A, Costagliola D, Haïk S. Susceptibility to Creutzfeldt-Jakob disease after human growth hormone treatment in France. Neurology. 2018 Aug 21. 91 (8):e724-e731. [QxMD MEDLINE Link].
Sakudo A. Inactivation Methods for Prions. Curr Issues Mol Biol. 2020. 36:23-32. [QxMD MEDLINE Link].
Brodbelt AR, Vinten J, Larkin S. Informing patient contacts about iatrogenic Creutzfeldt-Jakob disease. J Hosp Infect. 2020 Jun. 105 (2):325-328. [QxMD MEDLINE Link].
Belay ED, Gambetti P, Schonberger LB, et al. Creutzfeldt-Jakob disease in unusually young patients who consumed venison. Arch Neurol. 2001 Oct. 58(10):1673-8. [QxMD MEDLINE Link].
Raymond GJ, Bossers A, Raymond LD, et al. Evidence of a molecular barrier limiting susceptibility of humans, cattle and sheep to chronic wasting disease. EMBO J. 2000 Sep 1. 19(17):4425-30. [QxMD MEDLINE Link]. [Full Text].
Angers RC, Browning SR, Seward TS, et al. Prions in skeletal muscles of deer with chronic wasting disease. Science. 2006 Feb 24. 311(5764):1117. [QxMD MEDLINE Link].
Marsh RF, Kincaid AE, Bessen RA, Bartz JC. Interspecies transmission of chronic wasting disease prions to squirrel monkeys (Saimiri sciureus). J Virol. 2005 Nov. 79(21):13794-6. [QxMD MEDLINE Link].
Brandel JP, Vlaicu MB, Culeux A, Belondrade M, Bougard D, Grznarova K, et al. Variant Creutzfeldt-Jakob Disease Diagnosed 7.5 Years after Occupational Exposure. N Engl J Med. 2020 Jul 2. 383 (1):83-85. [QxMD MEDLINE Link].
Hamaguchi T, Noguchi-Shinohara M, Nozaki I, Nakamura Y, Sato T, Kitamoto T. Medical procedures and risk for sporadic Creutzfeldt-Jakob disease, Japan, 1999-2008. Emerg Infect Dis. 2009 Feb. 15(2):265-71. [QxMD MEDLINE Link]. [Full Text].
Satoh K, Fuse T, Nonaka T, Dong T, Takao M, Nakagaki T, et al. Postmortem Quantitative Analysis of Prion Seeding Activity in the Digestive System. Molecules. 2019 Dec 16. 24 (24):[QxMD MEDLINE Link].
Orrú CD, Yuan J, Appleby BS, Li B, Li Y, Winner D, et al. Prion seeding activity and infectivity in skin samples from patients with sporadic Creutzfeldt-Jakob disease. Sci Transl Med. 2017 Nov 22. 9 (417):[QxMD MEDLINE Link].
Martheswaran T, Desautels JD, Moshirfar M, Shmunes KM, Ronquillo YC, Hoopes PC. A Contemporary Risk Analysis of Iatrogenic Transmission of Creutzfeldt-Jakob Disease (CJD) via Corneal Transplantation in the United States. Ophthalmol Ther. 2020 Sep. 9 (3):465-483. [QxMD MEDLINE Link].
Appleby BS, Rincon-Beardsley TD, Appleby KK, Crain BJ, Wallin MT. Initial diagnoses of patients ultimately diagnosed with prion disease. J Alzheimers Dis. 2014 Jan 1. 42(3):833-9. [QxMD MEDLINE Link].
Seipelt M, Zerr I, Nau R, et al. Hashimoto's encephalitis as a differential diagnosis of Creutzfeldt-Jakob disease. J Neurol Neurosurg Psychiatry. 1999 Feb. 66(2):172-6. [QxMD MEDLINE Link].
Castillo P, Woodruff B, Caselli R, et al. Steroid-responsive encephalopathy associated with autoimmune thyroiditis. Arch Neurol. 2006 Feb. 63(2):197-202. [QxMD MEDLINE Link].
Cossu G, Melis M, Molari A, et al. Creutzfeldt-Jakob disease associated with high titer of antithyroid autoantibodies: case report and literature review. Neurol Sci. 2003 Oct. 24(3):138-40. [QxMD MEDLINE Link].
Mead S, Rudge P. CJD mimics and chameleons. Pract Neurol. 2017 Apr. 17 (2):113-121. [QxMD MEDLINE Link].
Moda F, Bolognesi ML, Legname G. Novel screening approaches for human prion diseases drug discovery. Expert Opin Drug Discov. 2019 Oct. 14 (10):983-993. [QxMD MEDLINE Link].
Green AJE. RT-QuIC: a new test for sporadic CJD. Pract Neurol. 2019 Feb. 19 (1):49-55. [QxMD MEDLINE Link].
Rhoads DD, Wrona A, Foutz A, Blevins J, Glisic K, Person M, et al. Diagnosis of prion diseases by RT-QuIC results in improved surveillance. Neurology. 2020 Aug 25. 95 (8):e1017-e1026. [QxMD MEDLINE Link].
Llorens F, Kruse N, Karch A, Schmitz M, Zafar S, Gotzmann N, et al. Validation of α-Synuclein as a CSF Biomarker for Sporadic Creutzfeldt-Jakob Disease. Mol Neurobiol. 2018 Mar. 55 (3):2249-2257. [QxMD MEDLINE Link].
Llorens F, Villar-Piqué A, Hermann P, Schmitz M, Goebel S, Waniek K, et al. Cerebrospinal fluid non-phosphorylated tau in the differential diagnosis of Creutzfeldt-Jakob disease: a comparative prospective study with 14-3-3. J Neurol. 2020 Feb. 267 (2):543-550. [QxMD MEDLINE Link].
Abu-Rumeileh S, Baiardi S, Polischi B, Mammana A, Franceschini A, Green A, et al. Diagnostic value of surrogate CSF biomarkers for Creutzfeldt-Jakob disease in the era of RT-QuIC. J Neurol. 2019 Dec. 266 (12):3136-3143. [QxMD MEDLINE Link].
Blennow K, Diaz-Lucena D, Zetterberg H, Villar-Pique A, Karch A, Vidal E, et al. CSF neurogranin as a neuronal damage marker in CJD: a comparative study with AD. J Neurol Neurosurg Psychiatry. 2019 Aug. 90 (8):846-853. [QxMD MEDLINE Link].
Zerr I, Villar-Piqué A, Schmitz VE, Poleggi A, Pocchiari M, Sánchez-Valle R, et al. Evaluation of Human Cerebrospinal Fluid Malate Dehydrogenase 1 as a Marker in Genetic Prion Disease Patients. Biomolecules. 2019 Nov 28. 9 (12):[QxMD MEDLINE Link].
Young GS, Geschwind MD, Fischbein NJ, et al. Diffusion-weighted and fluid-attenuated inversion recovery imaging in Creutzfeldt-Jakob disease: high sensitivity and specificity for diagnosis. AJNR Am J Neuroradiol. 2005 Jun-Jul. 26(6):1551-62. [QxMD MEDLINE Link].
Bizzi A, Pascuzzo R, Blevins J, Grisoli M, Lodi R, Moscatelli MEM, et al. Evaluation of a New Criterion for Detecting Prion Disease With Diffusion Magnetic Resonance Imaging. JAMA Neurol. 2020 Sep 1. 77 (9):1141-1149. [QxMD MEDLINE Link].
Renard D, Castelnovo G, Collombier L, Thouvenot E, Boudousq V. FDG-PET in Creutzfeldt-Jakob disease: Analysis of clinical-PET correlation. Prion. 2017 Nov 2. 11 (6):440-453. [QxMD MEDLINE Link].
Abu-Rumeileh S, Redaelli V, Baiardi S, Mackenzie G, Windl O, Ritchie DL, et al. Sporadic Fatal Insomnia in Europe: Phenotypic Features and Diagnostic Challenges. Ann Neurol. 2018 Sep. 84 (3):347-360. [QxMD MEDLINE Link].
Jones E, Hummerich H, Viré E, Uphill J, Dimitriadis A, et al. Identification of novel risk loci and causal insights for sporadic Creutzfeldt-Jakob disease: a genome-wide association study. Lancet Neurol. 2020 Oct. 19 (10):840-848. [QxMD MEDLINE Link].
Rees HC, Maddison BC, Owen JP, Whitelam GC, Gough KC. Concentration of disease-associated prion protein with silicon dioxide. Mol Biotechnol. 2009 Mar. 41(3):254-62. [QxMD MEDLINE Link].
Laude H. Beringue V. Newly discovered forms of prion diseases in ruminants. [Review]. Pathologie Biologie. 2009. 57(2):117-26.
Sim VL. Caughey B. Recent advances in prion chemotherapeutics. [Review]. Infectious Disorders - Drug Targets. 2009. 9(1):81-91.
Relano-Gines A. Gabelle A. Lehmann S. Milhavet O. Crozet C. Gene and cell therapy for prion diseases. [Review]. Infectious Disorders - Drug Targets. 2009. 9(1):58-68.
Miguelez-Rodriguez A, Santos-Juanes J, Vicente-Etxenausia I, Perez de Heredia-Goñi K, Garcia B, Quiros LM, et al. Brains with sporadic Creutzfeldt-Jakob disease and copathology showed a prolonged end-stage of disease. J Clin Pathol. 2018 May. 71 (5):446-450. [QxMD MEDLINE Link].
Caspi S, Halimi M, Yanai A, Sasson SB, Taraboulos A, Gabizon R. The anti-prion activity of Congo red. Putative mechanism. J Biol Chem. 1998 Feb 6. 273(6):3484-9. [QxMD MEDLINE Link].
Demaimay R, Harper J, Gordon H, Weaver D, Chesebro B, Caughey B. Structural aspects of Congo red as an inhibitor of protease-resistant prion protein formation. J Neurochem. 1998 Dec. 71(6):2534-41. [QxMD MEDLINE Link].
Doh-ura K, Ishikawa K, Murakami-Kubo I, et al. Treatment of transmissible spongiform encephalopathy by intraventricular drug infusion in animal models. J Virol. 2004 May. 78(10):4999-5006. [QxMD MEDLINE Link]. [Full Text].
Demaimay R, Chesebro B, Caughey B. Inhibition of formation of protease-resistant prion protein by Trypan Blue, Sirius Red and other Congo Red analogs. Arch Virol Suppl. 2000. 277-83. [QxMD MEDLINE Link].
Tagliavini F, McArthur RA, Canciani B, et al. Effectiveness of anthracycline against experimental prion disease in Syrian hamsters. Science. 1997 May 16. 276(5315):1119-22. [QxMD MEDLINE Link].
Adjou KT, Demaimay R, Lasmezas CI, Seman M, Deslys JP, Dormont D. Differential effects of a new amphotericin B derivative, MS-8209, on mouse BSE and scrapie: implications for the mechanism of action of polyene antibiotics. Res Virol. 1996 Jul-Aug. 147(4):213-8. [QxMD MEDLINE Link].
Adjou KT, Demaimay R, Deslys JP, et al. MS-8209, a water-soluble amphotericin B derivative, affects both scrapie agent replication and PrPres accumulation in Syrian hamster scrapie. J Gen Virol. 1999 Apr. 80 ( Pt 4):1079-85. [QxMD MEDLINE Link].
Adjou KT, Privat N, Demart S, et al. MS-8209, an amphotericin B analogue, delays the appearance of spongiosis, astrogliosis and PrPres accumulation in the brain of scrapie-infected hamsters. J Comp Pathol. 2000 Jan. 122(1):3-8. [QxMD MEDLINE Link].
Mange A, Milhavet O, McMahon HE, Casanova D, Lehmann S. Effect of amphotericin B on wild-type and mutated prion proteins in cultured cells: putative mechanism of action in transmissible spongiform encephalopathies. J Neurochem. 2000 Feb. 74(2):754-62. [QxMD MEDLINE Link].
Mange A, Nishida N, Milhavet O, McMahon HE, Casanova D, Lehmann S. Amphotericin B inhibits the generation of the scrapie isoform of the prion protein in infected cultures. J Virol. 2000 Apr. 74(7):3135-40. [QxMD MEDLINE Link].
Farquhar C, Dickinson A, Bruce M. Prophylactic potential of pentosan polysulphate in transmissible spongiform encephalopathies. Lancet. 1999 Jan 9. 353(9147):117. [QxMD MEDLINE Link].
Ladogana A, Casaccia P, Ingrosso L, et al. Sulphate polyanions prolong the incubation period of scrapie-infected hamsters. J Gen Virol. 1992 Mar. 73 ( Pt 3):661-5. [QxMD MEDLINE Link].
Farquhar CF, Dickinson AG. Prolongation of scrapie incubation period by an injection of dextran sulphate 500 within the month before or after infection. J Gen Virol. 1986 Mar. 67 ( Pt 3):463-73. [QxMD MEDLINE Link].
Priola SA, Raines A, Caughey WS. Porphyrin and phthalocyanine antiscrapie compounds. Science. 2000 Feb 25. 287(5457):1503-6. [QxMD MEDLINE Link].
Priola SA, Raines A, Caughey W. Prophylactic and therapeutic effects of phthalocyanine tetrasulfonate in scrapie-infected mice. J Infect Dis. 2003 Sep 1. 188(5):699-705. [QxMD MEDLINE Link].
Masullo C, Macchi G, Xi YG, Pocchiari M. Failure to ameliorate Creutzfeldt-Jakob disease with amphotericin B therapy. J Infect Dis. 1992 Apr. 165(4):784-5. [QxMD MEDLINE Link].
Korth C, May BC, Cohen FE, Prusiner SB. Acridine and phenothiazine derivatives as pharmacotherapeutics for prion disease. Proc Natl Acad Sci U S A. 2001 Aug 14. 98(17):9836-41. [QxMD MEDLINE Link]. [Full Text].
Nakajima M, Yamada T, Kusuhara T, et al. Results of quinacrine administration to patients with Creutzfeldt-Jakob disease. Dement Geriatr Cogn Disord. 2004. 17(3):158-63. [QxMD MEDLINE Link].
Barret A, Tagliavini F, Forloni G, et al. Evaluation of quinacrine treatment for prion diseases. J Virol. 2003 Aug. 77(15):8462-9. [QxMD MEDLINE Link].
Collins SJ, Lewis V, Brazier M, Hill AF, Fletcher A, Masters CL. Quinacrine does not prolong survival in a murine Creutzfeldt-Jakob disease model. Ann Neurol. 2002 Oct. 52(4):503-6. [QxMD MEDLINE Link].
Todd NV, Morrow J, Doh-ura K, et al. Cerebroventricular infusion of pentosan polysulphate in human variant Creutzfeldt-Jakob disease. J Infect. 2005 Jun. 50(5):394-6. [QxMD MEDLINE Link].
Soto C, Kascsak RJ, Saborio GP, et al. Reversion of prion protein conformational changes by synthetic beta-sheet breaker peptides. Lancet. 2000 Jan 15. 355(9199):192-7. [QxMD MEDLINE Link].
Wisniewski T, Aucouturier P, Soto C, Frangione B. The prionoses and other conformational disorders. Amyloid. 1998 Sep. 5(3):212-24. [QxMD MEDLINE Link].
Wisniewski T, Sigurdsson EM, Aucouturier P, et al. Conformation as a therapeutic target in the prionoses and other neurodegenerative conditions. Molecular and Cellular Pathology in Prion Disease. 2001.
De Gioia L, Selvaggini C, Ghibaudi E, et al. Conformational polymorphism of the amyloidogenic and neurotoxic peptide homologous to residues 106-126 of the prion protein. J Biol Chem. 1994 Mar 18. 269(11):7859-62. [QxMD MEDLINE Link].
Nguyen J, Baldwin MA, Cohen FE, Prusiner SB. Prion protein peptides induce alpha-helix to beta-sheet conformational transitions. Biochemistry. 1995 Apr 4. 34(13):4186-92. [QxMD MEDLINE Link].
Zhang H, Kaneko K, Nguyen JT, et al. Conformational transitions in peptides containing two putative alpha-helices of the prion protein. J Mol Biol. 1995 Jul 21. 250(4):514-26. [QxMD MEDLINE Link].
Naiki H, Higuchi K, Nakakuki K, Takeda T. Kinetic analysis of amyloid fibril polymerization in vitro. Lab Invest. 1991 Jul. 65(1):104-10. [QxMD MEDLINE Link].
Wisniewski T, Castano EM, Golabek A, Vogel T, Frangione B. Acceleration of Alzheimer's fibril formation by apolipoprotein E in vitro. Am J Pathol. 1994 Nov. 145(5):1030-5. [QxMD MEDLINE Link].
Sigurdsson EM, Permanne B, Soto C, Wisniewski T, Frangione B. In vivo reversal of amyloid-beta lesions in rat brain. J Neuropathol Exp Neurol. 2000 Jan. 59(1):11-7. [QxMD MEDLINE Link].
Soto C, Sigurdsson EM, Morelli L, Kumar RA, Castano EM, Frangione B. Beta-sheet breaker peptides inhibit fibrillogenesis in a rat brain model of amyloidosis: implications for Alzheimer's therapy. Nat Med. 1998 Jul. 4(7):822-6. [QxMD MEDLINE Link].
Vidal R, Ghiso J, Wisniewski T, Frangione B. Alzheimer's presenilin 1 gene expression in platelets and megakaryocytes. Identification of a novel splice variant. FEBS Lett. 1996 Sep 9. 393(1):19-23. [QxMD MEDLINE Link].
Sigurdsson EM, Brown DR, Alim MA, Scholtzova H, Carp R, Meeker HC, et al. Copper chelation delays the onset of prion disease. J Biol Chem. 2003 Nov 21. 278(47):46199-202. [QxMD MEDLINE Link].
Sigurdsson EM. Immunotherapy for conformational diseases. Curr Pharm Des. 2006. 12(20):2569-85. [QxMD MEDLINE Link].
Schenk D, Barbour R, Dunn W, et al. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature. 1999 Jul 8. 400(6740):173-7. [QxMD MEDLINE Link].
Weiner HL, Lemere CA, Maron R, et al. Nasal administration of amyloid-beta peptide decreases cerebral amyloid burden in a mouse model of Alzheimer's disease. Ann Neurol. 2000 Oct. 48(4):567-79. [QxMD MEDLINE Link].
Janus C, Pearson J, McLaurin J, et al. A beta peptide immunization reduces behavioural impairment and plaques in a model of Alzheimer's disease. Nature. 2000 Dec 21-28. 408(6815):979-82. [QxMD MEDLINE Link].
Morgan D, Diamond DM, Gottschall PE, et al. Arendash GW Ab peptide vaccination prevents memory loss in an animal model of Alzheimer's disease. Nature. 2001. 408:982-985.
Bard F, Cannon C, Barbour R, et al. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med. 2000 Aug. 6(8):916-9. [QxMD MEDLINE Link].
Sigurdsson EM, Brown DR, Daniels M, et al. Immunization delays the onset of prion disease in mice. Am J Pathol. 2002 Jul. 161(1):13-7. [QxMD MEDLINE Link].
Goni F, Knudsen E, Schreiber F, et al. Mucosal vaccination delays or prevents prion infection via an oral route. Neuroscience. 2005. 133(2):413-21. [QxMD MEDLINE Link].
Pankiewicz J, Sadowski M, Kascsak R, et al. Therapeutic mechanisms of anti-prion antibodies in a tissue culture model of prion infection. Soc Neurosci Abst. 2005.
Sigurdsson EM, Sy MS, Li R, Scholtzova H, Kascsak RJ, Kascsak R, et al. Anti-prion antibodies for prophylaxis following prion exposure in mice. Neurosci Lett. 2003 Jan 23. 336(3):185-7. [QxMD MEDLINE Link].
Abdelaziz DH, Thapa S, Abdulrahman B, Vankuppeveld L, Schatzl HM. Metformin reduces prion infection in neuronal cells by enhancing autophagy. Biochem Biophys Res Commun. 2020 Mar 5. 523 (2):423-428. [QxMD MEDLINE Link].
Manuelidis L. Vaccination with an attenuated Creutzfeldt-Jakob disease strain prevents expression of a virulent agent. Proc Natl Acad Sci U S A. 1998 Mar 3. 95(5):2520-5. [QxMD MEDLINE Link].
White AR, Enever P, Tayebi M, et al. Monoclonal antibodies inhibit prion replication and delay the development of prion disease. Nature. 2003 Mar 6. 422(6927):80-3. [QxMD MEDLINE Link].
Enari M, Flechsig E, Weissmann C. Scrapie prion protein accumulation by scrapie-infected neuroblastoma cells abrogated by exposure to a prion protein antibody. Proc Natl Acad Sci U S A. 2001 Jul 31. 98(16):9295-9. [QxMD MEDLINE Link]. [Full Text].
Peretz D, Williamson RA, Kaneko K, et al. Antibodies inhibit prion propagation and clear cell cultures of prion infectivity. Nature. 2001 Aug 16. 412(6848):739-43. [QxMD MEDLINE Link].
Souan L, Tal Y, Felling Y, Cohen IR, Taraboulos A, Mor F. Modulation of proteinase-K resistant prion protein by prion peptide immunization. Eur J Immunol. 2001 Aug. 31(8):2338-46. [QxMD MEDLINE Link].
Sigurdsson EM, Scholtzova H, Mehta PD, Frangione B, Wisniewski T. Immunization with a nontoxic/nonfibrillar amyloid-beta homologous peptide reduces Alzheimer's disease-associated pathology in transgenic mice. Am J Pathol. 2001 Aug. 159(2):439-47. [QxMD MEDLINE Link].
Sigurdsson EM, Knudsen E, Asuni A, Fitzer-Attas C, Sage D, Quartermain D, et al. An attenuated immune response is sufficient to enhance cognition in an Alzheimer's disease mouse model immunized with amyloid-beta derivatives. J Neurosci. 2004 Jul 14. 24(28):6277-82. [QxMD MEDLINE Link].
Aguzzi A, Frontzek K. New paradigms of clinical trial design for genetic prion diseases. Lancet Neurol. 2020 Apr. 19 (4):284-285. [QxMD MEDLINE Link].
Appleby BS, Yobs DR. Symptomatic treatment, care, and support of CJD patients. Handb Clin Neurol. 2018. 153:399-408. [QxMD MEDLINE Link].
Coste J. Prowse C. Eglin R. Fang C. Subgroup on TSE. A report on transmissible spongiform encephalopathies and transfusion safety. [Review]. Vox Sanguinis. 2009. 96(4):284-291.
Lefrere JJ, Hewitt P. From mad cows to sensible blood transfusion: the risk of prion transmission by labile blood components in the United Kingdom and in France. Transfusion. 2009 Apr. 49(4):797-812. [QxMD MEDLINE Link].
Brown P, Wolff A, Gajdusek DC. A simple and effective method for inactivating virus infectivity in formalin-fixed tissue samples from patients with Creutzfeldt-Jakob disease. Neurology. 1990 Jun. 40(6):887-90. [QxMD MEDLINE Link].
Ironside JW, Bell JE. The 'high-risk' neuropathological autopsy in AIDS and Creutzfeldt-Jakob disease: principles and practice. Neuropathol Appl Neurobiol. 1996 Oct. 22(5):388-93. [QxMD MEDLINE Link].
Aucouturier P, Kascsak RJ, Frangione B, Wisniewski T. Biochemical and conformational variability of human prion strains in sporadic Creutzfeldt-Jakob disease. Neurosci Lett. 1999 Oct 15. 274(1):33-6. [QxMD MEDLINE Link].
Brown P, Rau EH, Johnson BK, Bacote AE, Gibbs CJ Jr, Gajdusek DC. New studies on the heat resistance of hamster-adapted scrapie agent: threshold survival after ashing at 600 degrees C suggests an inorganic template of replication. Proc Natl Acad Sci U S A. 2000 Mar 28. 97(7):3418-21. [QxMD MEDLINE Link].
Budka H, Aguzzi A, Brown P, et al. Tissue handling in suspected Creutzfeldt-Jakob disease (CJD) and other human spongiform encephalopathies (prion diseases). Brain Pathol. 1995 Jul. 5(3):319-22. [QxMD MEDLINE Link].
Bueler H, Fischer M, Lang Y, et al. Normal development and behaviour of mice lacking the neuronal cell-surface PrP protein. Nature. 1992 Apr 16. 356(6370):577-82. [QxMD MEDLINE Link].
Castilla J, Saa P, Soto C. Detection of prions in blood. Nat Med. 2005 Sep. 11(9):982-5. [QxMD MEDLINE Link].
Chang B, Cheng X, Pan T, et al. An ultra-sensitive assay for detecting prions in blood. Proc Natl Acad Sci (USA) in press. 2006.
Citron M, Vigo-Pelfrey C, Teplow DB, et al. Excessive production of amyloid beta-protein by peripheral cells of symptomatic and presymptomatic patients carrying the Swedish familial Alzheimer disease mutation. Proc Natl Acad Sci U S A. 1994 Dec 6. 91(25):11993-7. [QxMD MEDLINE Link]. [Full Text].
Collinge J, Palmer MS, Dryden AJ. Genetic predisposition to iatrogenic Creutzfeldt-Jakob disease. Lancet. 1991 Jun 15. 337(8755):1441-2. [QxMD MEDLINE Link].
Collins SJ, Lawson VA, Masters CL. Transmissible spongiform encephalopathies. Lancet. 2004 Jan 3. 363(9402):51-61. [QxMD MEDLINE Link].
Dlouhy SR, Hsiao K, Farlow MR, et al. Linkage of the Indiana kindred of Gerstmann-Sträussler-Scheinker disease to the prion protein gene. Nat Genet. 1992 Apr. 1(1):64-7. [QxMD MEDLINE Link].
Dominguez DI, De Strooper B, Annaert W. Secretases as therapeutic targets for the treatment of Alzheimer's disease. Amyloid. 2001 Jun. 8(2):124-42. [QxMD MEDLINE Link].
Dropcho EJ. Update on paraneoplastic syndromes. Curr Opin Neurol. 2005 Jun. 18(3):331-6. [QxMD MEDLINE Link].
Finkenstaedt M, Szudra A, Zerr I, et al. MR imaging of Creutzfeldt-Jakob disease. Radiology. 1996 Jun. 199(3):793-8. [QxMD MEDLINE Link].
Foster PR. Prions and blood products. Ann Med. 2000 Oct. 32(7):501-13. [QxMD MEDLINE Link].
Gabizon R, Rosenmann H, Meiner Z, et al. Mutation and polymorphism of the prion protein gene in Libyan Jews with Creutzfeldt-Jakob disease (CJD). Am J Hum Genet. 1993 Oct. 53(4):828-35. [QxMD MEDLINE Link].
Goldfarb LG, Brown P, Goldgaber D, et al. Creutzfeldt-Jakob disease and kuru patients lack a mutation consistently found in the Gerstmann-Sträussler-Scheinker syndrome. Exp Neurol. 1990 Jun. 108(3):247-50. [QxMD MEDLINE Link].
Goldfarb LG, Brown P, McCombie WR, et al. Transmissible familial Creutzfeldt-Jakob disease associated with five, seven, and eight extra octapeptide coding repeats in the PRNP gene. Proc Natl Acad Sci U S A. 1991 Dec 1. 88(23):10926-30. [QxMD MEDLINE Link]. [Full Text].
Goldfarb LG, Haltia M, Brown P, et al. New mutation in scrapie amyloid precursor gene (at codon 178) in Finnish Creutzfeldt-Jakob kindred. Lancet. 1991 Feb 16. 337(8738):425. [QxMD MEDLINE Link].
Goldfarb LG, Mitrova E, Brown P, Toh BK, Gajdusek DC. Mutation in codon 200 of scrapie amyloid protein gene in two clusters of Creutzfeldt-Jakob disease in Slovakia. Lancet. 1990 Aug 25. 336(8713):514-5. [QxMD MEDLINE Link].
Goldfarb LG, Petersen RB, Tabaton M, et al. Fatal familial insomnia and familial Creutzfeldt-Jakob disease: disease phenotype determined by a DNA polymorphism. Science. 1992 Oct 30. 258(5083):806-8. [QxMD MEDLINE Link].
Goldgaber D, Goldfarb LG, Brown P, et al. Mutations in familial Creutzfeldt-Jakob disease and Gerstmann-Sträussler-Scheinker's syndrome. Exp Neurol. 1989 Nov. 106(2):204-6. [QxMD MEDLINE Link].
Haass C, Hung AY, Schlossmacher MG, Teplow DB, Selkoe DJ. beta-Amyloid peptide and a 3-kDa fragment are derived by distinct cellular mechanisms. J Biol Chem. 1993 Feb 15. 268(5):3021-4. [QxMD MEDLINE Link].
Head MW, Bunn TJ, Bishop MT, et al. Prion protein heterogeneity in sporadic but not variant Creutzfeldt-Jakob disease: UK cases 1991-2002. Ann Neurol. 2004 Jun. 55(6):851-9. [QxMD MEDLINE Link].
Hetz C, Soto C. Protein misfolding and disease: the case of prion disorders. Cell Mol Life Sci. 2003 Jan. 60(1):133-43. [QxMD MEDLINE Link].
Hill AF, Zeidler M, Ironside J, Collinge J. Diagnosis of new variant Creutzfeldt-Jakob disease by tonsil biopsy. Lancet. 1997 Jan 11. 349(9045):99-100. [QxMD MEDLINE Link].
Hsiao K, Baker HF, Crow TJ, et al. Linkage of a prion protein missense variant to Gerstmann-Sträussler syndrome. Nature. 1989 Mar 23. 338(6213):342-5. [QxMD MEDLINE Link].
Hsiao K, Dlouhy SR, Farlow MR, et al. Mutant prion proteins in Gerstmann-Sträussler-Scheinker disease with neurofibrillary tangles. Nat Genet. 1992 Apr. 1(1):68-71. [QxMD MEDLINE Link].
Hsiao K, Doh-ura K, KitamotoT. A prion protein amino acid substitution in ataxic Gerstmann-Straussler syndrome. Ann Neurol. 1989. 26:137.
Hsiao K, Meiner Z, Kahana E, et al. Mutation of the prion protein in Libyan Jews with Creutzfeldt-Jakob disease. N Engl J Med. 1991 Apr 18. 324(16):1091-7. [QxMD MEDLINE Link].
Jeong JK, Lee JH, Moon JH, Lee YJ, Park SY. Melatonin-mediated ß-catenin activation protects neuron cells against prion protein-induced neurotoxicity. J Pineal Res. 2014 Nov. 57(4):427-34. [QxMD MEDLINE Link].
Kitamoto T, Ohta M, Doh-ura K, Hitoshi S, Terao Y, Tateishi J. Novel missense variants of prion protein in Creutzfeldt-Jakob disease or Gerstmann-Sträussler syndrome. Biochem Biophys Res Commun. 1993 Mar 15. 191(2):709-14. [QxMD MEDLINE Link].
Kovacs GG, Voigtlander T, Gelpi E, Budka H. Rationale for diagnosing human prion disease. World J Biol Psychiatry. 2004 Apr. 5(2):83-91. [QxMD MEDLINE Link].
Maness LM, Banks WA, Podlisny MB, Selkoe DJ, Kastin AJ. Passage of human amyloid beta-protein 1-40 across the murine blood-brain barrier. Life Sci. 1994. 55(21):1643-50. [QxMD MEDLINE Link].
Miyazono M, Kitamoto T, Doh-ura K, Iwaki T, Tateishi J. Creutzfeldt-Jakob disease with codon 129 polymorphism (valine): a comparative study of patients with codon 102 point mutation or without mutations. Acta Neuropathol. 1992. 84(4):349-54. [QxMD MEDLINE Link].
Nochlin D, Sumi SM, Bird TD, et al. Familial dementia with PrP-positive amyloid plaques: a variant of Gerstmann-Sträussler syndrome. Neurology. 1989 Jul. 39(7):910-8. [QxMD MEDLINE Link].
Owen F, Poulter M, Lofthouse R, et al. Insertion in prion protein gene in familial Creutzfeldt-Jakob disease. Lancet. 1989 Jan 7. 1(8628):51-2. [QxMD MEDLINE Link].
Owen F, Poulter M, Shah T, et al. An in-frame insertion in the prion protein gene in familial Creutzfeldt-Jakob disease. Brain Res Mol Brain Res. 1990 Apr. 7(3):273-6. [QxMD MEDLINE Link].
Quadrio I, Ugnon-Cafe S, Dupin M, Esposito G, Streichenberger N, Krolak-Salmon P. Rapid diagnosis of human prion disease using streptomycin with tonsil and brain tissues. Lab Invest. 2009 Apr. 89(4):406-13. [QxMD MEDLINE Link].
Ripoll L, Laplanche JL, Salzmann M, et al. A new point mutation in the prion protein gene at codon 210 in Creutzfeldt-Jakob disease. Neurology. 1993 Oct. 43(10):1934-8. [QxMD MEDLINE Link].
Sakaguchi S. Prospects for preventative vaccines against prion diseases. Protein & Peptide Letters. 2009. 16(3):260-70.
Soto C, Saborio GP, Anderes L. Cyclic amplification of protein misfolding: application to prion-related disorders and beyond. Trends Neurosci. 2002 Aug. 25(8):390-4. [QxMD MEDLINE Link].
Weissmann C. Molecular genetics of transmissible spongiform encephalopathies. J Biol Chem. 1999 Jan 1. 274(1):3-6. [QxMD MEDLINE Link].
Will RG, Ironside JW, Zeidler M, et al. A new variant of Creutzfeldt-Jakob disease in the UK. Lancet. 1996 Apr 6. 347(9006):921-5. [QxMD MEDLINE Link].
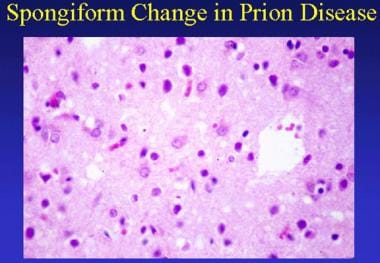 Prion-related diseases. Spongiform change in prion disease. This section shows mild parenchymal vacuolation and prominent reactive astrocytosis.
Prion-related diseases. Spongiform change in prion disease. This section shows mild parenchymal vacuolation and prominent reactive astrocytosis.




