Practice Essentials
Refsum disease and the hepatic porphyrias are rare inherited neurodegenerative conditions with exacerbations and remissions due to abnormal metabolism of large tetrapyrrole molecules. Two common examples of large tetrapyrrole molecules are chlorophyll a, the photosynthetic pigment of green plants, and heme, the prosthetic group of hemoglobin (see the image below). Side groups on both species involve relatively small organic groups (methyl, vinyl, and free propionyl); one major exception is phytol, a large hydrocarbon alcoholic substituent on chlorophyll. Patients in both disease categories must avoid foods and drugs that lead to high levels of the relevant biological toxin, which can trigger or perpetuate an exacerbation. Refsum disease is classified into 2 subgroups, based on differences in enzymes affected, metabolites accumulated, genetics, clinical presentation, and treatment: classic/adult Refsum disease and infantile Refsum disease. [1, 2, 3, 4, 5, 6]
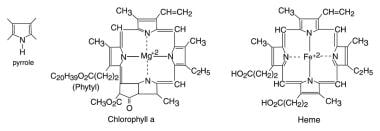 Tetrapyrrole molecules are large-ringed structures developed from 4 pyrrole groups and used in energy metabolism in both plants and animals.
Tetrapyrrole molecules are large-ringed structures developed from 4 pyrrole groups and used in energy metabolism in both plants and animals.
Heme is an iron-containing tetrapyrrole essential for diverse biological functions, including gas transport and sensing, oxidative metabolism, and xenobiotic detoxification. Heme is essential yet toxic in excess. As such, heme homeostasis is tightly regulated. [7]
The porphyrias are a set of metabolic disorders, each representing a defect in 1 of the 8 enzymes in the heme biosynthetic pathway that results in accumulation of organic compounds called porphyrins. This leads to the clinical and biochemical profile typical for each porphyria. Hepatic porphyrias are those in which enzyme deficiency occurs in the liver. Hepatic porphyrias include acute intermittent porphyria (AIP), variegate porphyria (VP), aminolevulinic acid dehydratase deficiency porphyria (ALAD), hereditary coproporphyria (HCP), and porphyria cutanea tarda (PCT). These conditions are distinct but have in common the accumulation of heme precursors. [8]
Testing and diagnosis
Establishing the diagnosis depends on clinical manifestations, biochemistry analysis of peroxisomal enzyme, phytanic acid concentration in plasma, and molecular genetic testing. [1] The biochemical hallmark of porphyrias is the overproduction of porphyrin precursors and porphyrin species. Afflicted patients present with myriad symptoms causing a diagnostic odyssey. Symptoms often overlap with those of common diseases and may be overlooked unless clinical suspicion is heightened. [9]
In acute disease (AHP), blood or urine tests are definitive, including increased phytanic acid in serum for Refsum disease; increased ALA and porphobilinogen (PBG) in the urine and serum for porphyrias; and low uroporphyrinogen decarboxylase [10] level in porphyria cutanea tarda. When porphyria is suspected in a patient without a family history, lead levels from blood or 24-hour urine collection should be obtained to exclude lead poisoning.
Once the biochemical tests indicate AHP, confirmation of the specific type of AHP is usually established by genetic testing, with sequencing of the 4 genes ALAD, HMBS, CPOX, and PPOX. When whole-gene sequencing is performed, 95-99% of cases can be identified. [3]
Electromyography should include a needle exam and nerve conduction exam for polyneuropathy. Refsum disease and the hepatic porphyrias are both associated with polyneuropathy, with both segmental and axonal characteristics.
Treatment
No specific treatments are indicated in Refsum disease, other than dietary restrictions of beef and milk products. Avoiding direct sunlight is necessary in preventing photosensitive dermatitis, especially in PCT. In Refsum disease, dietary intake of phytol and phytanic acid must be restricted.
Schedule annual or 6-month visits for a general physical examination. Analyze the patient's progress in avoiding exacerbating triggers, and order blood tests used to monitor adequate control (phytanic acid or ALA/PBG).
Givosiran (Givlaari) was approved by the FDA for adults with acute hepatic porphyrias (AHP), in which attacks are caused by induction of the enzyme 5-aminolevulinic acid synthase 1 (ALAS1). [11, 12, 13]
Gabapentin is useful as a long-term anticonvulsant in patients with hepatic porphyrias. It is the first drug of choice because it does not require hepatic metabolism; incidentally, it also is well tolerated by these patients in treatment of chronic pain, as an alternative to narcotics, which invoke liver metabolism.
Levetiracetam is a viable alternative to gabapentin if the side-effect profile (most notably somnolence) makes gabapentin undesirable. It does not provide a pain-reducing action like gabapentin.
Triple bromide is an alternative to the more traditional anticonvulsant choices of gabapentin and levetiracetam for long-term anticonvulsant therapy. Before the discovery of safe traditional anticonvulsants, it was the only treatment option for seizures in patients with hepatic porphyria. No specific dosing requirement is known, but the therapeutic range is 60-90 mg/dL to avoid toxic encephalopathy. Bromide preparation requires the assistance of a skilled pharmacist with compounding experience.
Because of its renal clearance, diphenhydramine is safe for use as a sleeping aid or antianxiety medication.
Treatment for the porphyrias consists of administration of intravenous hemin to prevent symptom progression, usually given once daily at a dose of 3-4 mg/kg body weight, typically for 4 days. [3] Liver transplant is reserved for patients with life-threatening acute attacks or progression of symptoms despite hemin therapy. [9] Early diagnosis is key for appropriate management and prevention of long-term complications in these rare disorders. [14]
Liver transplantation has been found to be curative in both the United States and Europe for patients with recurrent attacks refractory to treatment. [15, 16, 17]
Although a gastroenterologist or a physician with specific interest in porphyria may be helpful in planning disease management, a doctoral level clinical pharmacist or pharmacologist is especially helpful in making choices of safe drug combinations. Medical geneticists can help establish diagnostic histories and help to order the appropriate diagnostic tests, as well as provide genetic counseling.
AGA best practice advice
The American Gastroenterological Association (AGA) published the following best-practice-advice statements [3] :
-
Women aged 15-50 years with unexplained, recurrent severe abdominal pain without a clear etiology after an initial workup should be considered for screening for an AHP.
-
Initial diagnosis of AHP should be made by biochemical testing measuring δ-aminolevulinic acid, porphobilinogen, and creatinine on a random urine sample.
-
Genetic testing should be used to confirm the diagnosis of AHP in patients with positive biochemical testing.
-
Acute attacks of AHP that are severe enough to require hospital admission should be treated with intravenous hemin, given daily, preferably into a high-flow central vein.
-
In addition to intravenous hemin, management of acute attacks of AHP should include pain control, antiemetics, and management of systemic arterial hypertension, tachycardia, hyponatremia, and hypomagnesemia.
-
Patients should be counseled to avoid identifiable triggers that may precipitate acute attacks, such as alcohol and porphyrinogenic medications.
-
Prophylactic heme therapy or givosiran, administered in an outpatient setting, should be considered in patients with recurrent attacks (4 or more per year).
-
Liver transplantation for AHP should be limited to patients with intractable symptoms and significantly decreased quality of life who are refractory to pharmacotherapy.
-
Patients with AHP should be monitored annually for liver disease.
-
Patients with AHP, regardless of the severity of symptoms, should undergo surveillance for hepatocellular carcinoma, beginning at age 50 years, with liver ultrasound every 6 months.
-
Patients with AHP on treatment should undergo surveillance for chronic kidney disease annually with serum creatinine and estimated glomerular filtration rate.
-
Patients should be counseled on the chronic and long-term complications of AHP, including neuropathy, chronic kidney disease, hypertension, and hepatocellular carcinoma, as well as the need for long-term monitoring.
Pathophysiology
The neurotoxin in Refsum disease is phytanic acid, which in affected individuals is stored in neural and visceral parenchyma because of a deficiency in phytanic acid alpha-hydroxylase. The source of phytanic acid is either direct absorption or conversion of absorbed phytol from ruminant fat in meat or milk (only ruminants can release phytol from chlorophyll during digestion). Homozygosity is required for significant phytanic acid build-up.
The hepatic porphyrias are also associated with neurologic problems. The neurotoxins in these conditions are porphyrin precursors (delta-aminolevulinic acid [ALA], porphobilinogen [PBG]) and porphyrinogen substrates in heme synthesis, whose levels are elevated (see the image below). The actual porphyrins are oxidized products of the substrates, which are excreted in the feces and urine (the latter characterized by its reddish-brown, fluorescent color.)
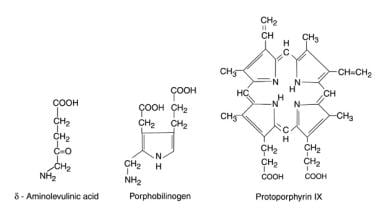
Whereas the enzyme deficiency in Refsum disease is inherited in an autosomal-recessive pattern, the enzyme deficiencies involved in the hepatic porphyrias typically are inherited in an autosomal-dominant mode. The hepatic porphyrias account for a varying spectrum of upstream accumulations of porphyrins and porphyrin precursors specific to each type of porphyria. [18] The following are common hepatic porphyrias:
-
Acute intermittent porphyria (AIP): Uroporphyrinogen synthase (or "porphobilinogen deaminase") deficiency with high ALA or PBG in urine and serum
-
Variegate porphyria (VP): Protoporphyrinogen oxidase deficiency [19] with high fecal levels of protoporphyrin and coproporphyrin [20]
-
Hereditary coproporphyria: Coproporphyrinogen oxidase deficiency with high urinary and/or fecal levels of coproporphyrins
-
Porphyria cutanea tarda (PCT): Uroporphyrinogen decarboxylase deficiency with high urinary and red cell levels of uroporphyrin
-
Erythropoietic protoporphyria (EPP), "protoporphyria", or "erythrohepatic protoporphyria" (not a hepatic porphyria) [21, 22] : Ferrochelatase deficiency, whereby fecal and erythrocyte levels of protoporphyrins are increased without any urinary porphyrins; abnormal porphyrin metabolism originates in erythrocytes, not the liver, yet ironically, patients with EPP may develop chronic liver failure; EPP is not characterized by neurologic symptoms and does not respond to sugar or hemin (ie, hematin) treatment
Epidemiology
Refsum disease is rare, but heterozygote carriers may be at risk if they have diets highly selective for beef and dairy products.
Acute intermittent porphyria (AIP) is the most common type of acute hepatic porphyria (AHP), with an estimated prevalence of patients with symptoms of approximately 1 in 100,000. [3] AIP is widely believed to be latent in 90% of cases. Porphyria cutanea tarda is believed to be the most common type, but because of poor recording, no data have been published. Erythropoietic protoporphyria is also believed to be common but not clearly documented. Other forms of true hepatic porphyrias are rare except variegate porphyria in individuals with ancestry of Afrikaner lineage. Variegate porphyria is common in South Africa (about 3 cases per 1000 population).
Mortality/Morbidity
Both Refsum disease and hepatic porphyrias are characterized by remissions and exacerbations of neurologic dysfunction, which can resolve completely or manifest stepwise deterioration. Permanent residual deficits are not uncommon; residual defects during latent periods include polyneuropathy in both conditions, ataxia and retinitis pigmentosa with night blindness in Refsum disease, [23] and photosensitive dermatitis in porphyrias (rare in acute intermittent porphyria). Death in either disease is commonly caused by cardiac arrhythmias during exacerbations. Cardiomyopathy can occur in Refsum disease beause of phytanic acid storage and in acute porphyric crises because of electrolyte disturbance (in as many as 25% of acute intermittent porphyria cases).
Race
Both Refsum disease and the porphyrias tend to occur more often in individuals of white hereditary lineage. The exception is porphyria cutanea tarda, which is noted among blacks of Bantu lineage (as well as whites).
Acute intermittent porphyria is most common among whites of English or Scandinavian heritage. Variegate porphyria is most common among Afrikaners, selectively concentrated in royal European lineage (eg, as documented in the popular film "The Madness Of King George"), and also present in certain large families of Great Britain, Holland, Sweden, and the United States.
Sex
Prevalence is expected to be equal between the sexes because of autosomal inheritance; however, clinical attacks may be more common in females with acute intermittent porphyria and in males with porphyria cutanea tarda. Consanguinity, causing the homozygous recessive condition, is not an uncommon cause of Refsum disease.
Age
Initial attacks in both disease categories can occur in early childhood, but in the hepatic porphyrias, the onset is usually postpubertal. [24] Erythropoietic protoporphyria is characterized by childhood onset of acute cutaneous photosensitivity to direct sunlight.
Childhood epilepsy is an exception to the postpuberty onset rule for initial porphyric attacks in the hepatic forms. Long-term drug use in idiopathic epilepsy with inactive or latent hepatic porphyria, even in prepubertal children, is a potent activator of cytochrome P450. Liver synthesis of heme groups is accelerated, leading to high levels of porphyrins and premature porphyric crises.
Earlier-onset Refsum disease is due to a pervasive dietary risk from consuming large quantities of beef and, to a greater degree, milk. For this same reason, persisting residual deficits are typical by age 20 years. Sporadic intake of provocative drugs in latent porphyria can induce exacerbations and eventually lead to persisting residual deficits.
Prognosis
Prognosis in Refsum disease and the hepatic porphyrias depends entirely on the proper dietary and drug restrictions.
The patient's ability to survive an acute exacerbation depends on the adequacy of acute care (especially the care available in an intensive care unit).
Survival for infantile Refsum disease is generally 5 to 13 years and maybe until adulthood. Survival for adult Refsum disease is the fourth to fifth decade of life. Prognosis is poor in untreated or noncompliant patients. Progressive degeneration of myelinated nerve fibers and of cardiac electro-conduction pathways leads to central and peripheral neuropathic symptoms, cardiac arrhythmias, impaired vision, and hearing loss. Arrhythmias are a frequent cause of death. [1]
Survival in an acute porphyric attack depends on monitoring levels of porphyrins and the proper use of D10W and hemin infusions. Despite appropriate treatment measures, the mortality rate in acute attacks of acute intermittent porphyria may be as high as 25%.
Patient Education
Both patients with Refsum disease and those with porphyria must become experts in understanding their disease. Both the Hereditary Disease Foundation and the American Porphyria Foundation can be a source of peer support and information for patients and physicians.
Monitoring of phytanic acid and ALA/PBG levels is a key element in providing outpatient treatment.
For patients with Refsum disease, specific review of safe dietary patterns must be a regular part of outpatient care.
Safety of specific drugs must be emphasized to the patient with porphyria (ie, avoiding drugs that induce cytochrome P450 activity). Encourage patients to seek advice by telephone if they have questions or concerns.
Genetic inheritance patterns must be understood, so that the patient can exercise responsibility in sexual relations or family planning.
Patients with porphyria have a 50% risk of passing along an autosomal-dominant trait with high expressivity.
Patients with Refsum disease should avoid marriage or sexual involvement with blood relatives (consanguinity), especially if distant relatives have diagnoses of neurodegenerative disease with childhood onset. Planning offspring is especially difficult, since no simple method is available to detect heterozygotes in this autosomal-recessive disease.
-
Tetrapyrrole molecules are large-ringed structures developed from 4 pyrrole groups and used in energy metabolism in both plants and animals.
-
Three characteristic substrate molecules of the heme porphyrin pathway.





