Background
Lesch-Nyhan disease is a genetic disorder associated with 3 major clinical elements: overproduction of uric acid, neurologic disability, and behavioral problems. [1] The overproduction of uric acid is associated with hyperuricemia. If left untreated, it can produce nephrolithiasis with renal failure, gouty arthritis, and solid subcutaneous deposits known as tophi. The neurologic disability is dominated by dystonia but may include choreoathetosis, ballismus, spasticity, or hyperreflexia. [2] The behavioral problems include intellectual disability and aggressive and impulsive behaviors. Patients with the classic disease also develop persistent and severe self-injurious behavior.
In addition to the classic clinical disease, patients with less severe disease and partial syndromes are increasingly recognized. [3] In these milder variants, self-injury may not occur, cognition may be normal, or dystonia may be mild or even absent. Some may have overproduction of uric acid and its consequences alone. These patients are identified by demonstrating HPRT deficiency or a mutation in the HPRT gene. Collectively, they are referred to as Lesch-Nyhan variants.
Treatment of the condition is limited. Allopurinol is useful to control the overproduction of uric acid and reduces the risk of nephrolithiasis and gouty arthritis. Few treatments have proven consistently helpful for the neurologic or behavioral difficulties. Motor disability is managed with a combination of baclofen and benzodiazepines, while the behavioral abnormalities are best managed by a combination of behavioral modification techniques and medications.
Pathophysiology
Hypoxanthine-guanine phosphoribosyl transferase (HPRT) normally plays a key role in the recycling of the purine bases, hypoxanthine and guanine, into the purine nucleotide pools (see the image below).
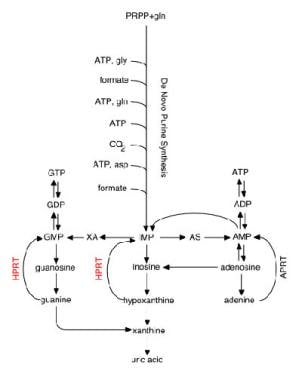
In the absence of HPRT, these purine bases cannot be salvaged; instead, they are degraded and excreted as uric acid. In addition to the failure of purine recycling, the synthetic rate for purines is accelerated, presumably to compensate for purines lost by the failure of the salvage process. The failure of recycling together with the increased synthesis of purines is the basis for the overproduction of uric acid. [1]
The increased production of uric acid leads to hyperuricemia. Since uric acid is near its physiologic limit of solubility in the body, the persistent hyperuricemia increases the risk of uric acid crystal precipitation in the tissues to form tophi. Uric acid crystal deposition in the joints produces an inflammatory reaction and gouty arthritis. The kidneys respond to the hyperuricemia by increasing its excretion into the urogenital system, increasing the risk of forming urate stones in the urinary collecting system. These stones may be passed as a sandy sludge or as larger particles that may obstruct urine flow and increase the risk of hematuria and urinary tract infections.
The pathogenesis of the neurologic and behavioral features is incompletely understood. [4] Neurochemical and neuroimaging studies have demonstrated significant abnormalities of dopamine neuron function in the basal ganglia that might account for the abnormal extrapyramidal neurologic signs and many of the behavioral anomalies. [5] Neuropathologic studies suggest a neurodevelopmental defect, with no signs of a degenerative process. [2] However, the mechanism by which HPRT deficiency influences the basal ganglia, and particularly the dopamine systems, remains unknown.
Etiology
Lesch-Nyhan disease and its variants are caused by mutations in the HPRT gene on the X chromosome. [6] The mutations are heterogeneous, with more than 600 different ones documented, including single base substitutions, deletions, insertions, or substitutions (see the image below). [7, 8]
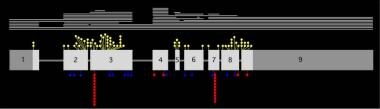 The HPRT gene has 9 exons, with the coding region depicted as light gray boxes. Genetic mutations in Lesch-Nyhan disease and its variants are heterogenous and include point mutations leading to amino acid substitution (yellow circles), point mutations leading to premature stop (red squares), insertions (blue triangles), deletions (white lines), and other more complex changes (not shown).
The HPRT gene has 9 exons, with the coding region depicted as light gray boxes. Genetic mutations in Lesch-Nyhan disease and its variants are heterogenous and include point mutations leading to amino acid substitution (yellow circles), point mutations leading to premature stop (red squares), insertions (blue triangles), deletions (white lines), and other more complex changes (not shown).
Epidemiology
The reported worldwide prevalence of Lesch-Nyhan disease is 1 case per 380,000 population. The disease has been reported in most races, with approximately equal rates for most ethnic groups. Few patients live beyond 40 years. [9]
Lesch-Nyhan disease is an X-linked recessive disorder; therefore, nearly all cases are in males. Only rarely has the disease been reported in females. [9]
Prognosis
With optimal medical care, individuals with Lesch-Nyhan disease typically live into their third or even fourth decade of life. Few patients live beyond 40 years.
Many patients die of aspiration pneumonia or complications from chronic nephrolithiasis and renal failure; however, a significant proportion of patients die suddenly and unexpectedly from unknown causes. [10]
Despite the use of allopurinol to control hyperuricemia, some patients still succumb to the consequences of recurrent nephrolithiasis, such as renal failure or urosepsis. Other patients experience progressive dysphagia and die after aspiration and pneumonia. Sudden, unexpected death also occurs, even on a background of an apparently stable medical condition. The reasons for sudden death remain unknown, though respiratory failure from cervical pathology or laryngospasm are considered leading possibilities. [10]
Patient Education
Since few reliable treatments are available for Lesch-Nyhan disease, genetic counseling is critical for prevention. Mothers and sisters of patients should be tested to determine if they are carriers. [11, 12]
Prenatal testing should be offered to all pregnant women known to be carriers. Because of the rare potential for gonadal mosaicism, prenatal testing should also be offered to mothers who have previously given birth to an affected individual, even if she does not appear to be a carrier.
-
Purine metabolic pathways.
-
A small portion of the lower lip has been disfigured by persistent self-biting.
-
The distal portions of several fingers are shortened by prior uncontrolled self-biting.
-
The HPRT gene has 9 exons, with the coding region depicted as light gray boxes. Genetic mutations in Lesch-Nyhan disease and its variants are heterogenous and include point mutations leading to amino acid substitution (yellow circles), point mutations leading to premature stop (red squares), insertions (blue triangles), deletions (white lines), and other more complex changes (not shown).





