Background
Epilepsy is a disease associated with a pathologic and enduring tendency to have recurrent seizures. Focal epilepsies, also termed partial or localization-related epilepsies, are seizure disorders that originate within a neuronal network limited to one hemisphere, whether unifocal or multifocal. This is in contrast to generalized epilesies, where seizures rapidly engage bilateral distributed networks.
Focal epilepsies represent the most common type of adult-onset epilepsy. By definition, there is focal abnormality that underlies focal epilepsies; while these structural abnormalities can sometimes be identified with imaging, they often remain of unknown etiology (eg, negative MRI). When a cause is found, it can include various structural lesions (eg, mesiotemporal sclerosis, traumatic scars, neoplasms, vascular malformations, strokes, neuronal heterotopias, etc.). In children, cortical dysplasias and low-grade neoplasms are the most commonly identified causes (see Etiology).
Obtaining a description of the seizures from the patient and any witnesses is critical. The description needs to include a description of the patient’s level of awareness during the seizure, the most prominent early motor and non-motor semiological features, and whether the seizure subsequently evolved into a bilateral tonic-clonic seizure (see Clinical Presentation). “Focal aware seizure” replaces the previous term “simple partial seizure” and a “focal impaired awareness seizure” replaces the term “complex partial seizure.”
Further evaluation, which may include neuroimaging, laboratory studies, and electroencephalography, is important for determining the specific disorder to determine prognosis and guide therapy (see Workup).
Focal epilepsies are generally treated with antiseizure medications (ASMs). Nonpharmacologic treatments in refractory cases include surgery, neurostimulation, and dietary modification (see Treatment and Management).
Go to Epilepsy and Seizures for a general overview of this topic.
Pathophysiology
Seizures may be understood as paroxysmal cerebral dysrhythmias, whereby there is a “transient occurrence of signs and symptoms due to abnormal excessive or synchronous activity in the brain.” [1]
In focal seizures, this pattern of transient electrical hypersynchrony begins in a circumscribed hyperexcitable neuronal population, termed the epileptic focus. An alteration in voltage-gated ion channels caused by decreased inhibitory and increased excitatory neurotransmission mediates this hyperexcitability. The pathological correlate on a neurophysiologic level for this aberrant electrical activity is embodied in the paroxysmal depolarization shift, whereby a cortical neuron undergoes a calcium-dependent depolarization followed by a prolonged hyperpolarization phase. If several million neurons discharge at once, the summated electrical potential may be seen on scalp EEG as a focal interictal epileptiform spike. This local aberrant activity may cause a cascade of hypersynchronous discharges throughout larger brain networks, culminating in the clinical and electrophysiological manifestation of a seizure. Therefore, while the epileptic focus may be restricted to a small focus, it is possible for it to secondarily involve generalized cerebral networks.
In turn, focal epilepsy represents a permanent alteration in the epileptic focus, a process termed epileptogenesis, such that the individual develops a propensity for repeated seizures. Epileptogenesis may be mediated by congenital (genetic conditions or migrational defects) or acquired processes (cerebral infarctions, neoplasms, or infections), although the underlying etiology is sometimes unknown.
Although the traditional understanding of focal epilepsy has focused on hypersynchrony precipitated by the epileptic focus alone, there is increasing recognition that patients with focal epilepsy suffer from widespread brain dysfunction. Over time, aberrant coupling between different neuronal populations that synchronously fire may potentiate the development of epileptic networks and new epileptogenic foci distant to the original focus. [2, 3, 4]
Etiology
In the 2017 International League of Epilepsy (ILAE) classification syndrome, epileptic etiologies are divided into six non-mutually exclusive categories: structural, genetic, infectious, metabolic, immune, and unknown. [5] It should be noted that the terms “symptomatic” and “cryptogenic” were confusing and are no longer used in the current classification scheme.
Most focal epilepsies do not have a clear cause visible on imaging. A structural etiology may be related to genetic causes, such as cortical malformations, or acquired, such as stroke, trauma, or infection. One of the most common structural correlates in surgically amenable temporal lobe epilepsy is hippocampal sclerosis, characterized by CA1 and CA4-predominant neuronal loss and associated epileptogenesis. [6] In children, cortical dysplasias are the most implicated structural lesions, while lesions related to cerebral infarction are the most common cause of focal epilepsy in the elderly. When a structural lesion has a well-defined etiology, one may use both etiological terms concomitantly. As an example, tuberous sclerosis, which may present with focal epilepsy secondary to cortical tubers, is caused by mutations in genes encoding hamartin and tuberin; in this case, it is acceptable to reference both structural and genetic etiologies.
A genetic etiology may be invoked if there is a known or inferred mutation that is thought to account for the focal epilepsy syndrome. Supporting evidence for a genetic etiology may be garnered based on familial inheritance patterns or molecular/twin research studies in patients with the same syndrome. It should be noted that a genetic etiology does not always imply an inherited condition, as de novo mutations are also possible. Owing to phenotypic and genetic heterogeneity, genetic focal epilepsies remain under heavy investigation. Autosomal dominant forms of frontal and temporal lobe epilepsies as well as familial focal epilepsy syndromes have well-described genetic associations (CHRNA2, CHRNA4, LGI1, DEPDC5, etc.) and are often related to dysregulation in the mechanistic target of rapamycin complex 1 (mTORC1) pathway. [7] Next-generation sequencing has also recently uncovered a possible genetic basis for the epilepsy–aphasia spectrum epilepsies, with up to 20% of patients with mutations on the NMDA glutamate receptor subunit gene GRIN2A. [8]
Infectious etiologies represent the most common worldwide etiology for focal epilepsies. This designation is only used to describe enduring epilepsies resulting from infectious causes and not acute seizures in the setting of infection. Infectious focal epilepsies may not always have a structural correlate on diagnostic workup.
Metabolic etiologies may overlap with genetic etiologies and often result in generalized epilepsy syndromes. However, there are some well-characterized focal epilepsy syndromes purported to result from structural defects induced by metabolic syndromes. As an example, the rare hepatocerebral disorder Alpers’ syndrome caused by POLG1 mutations is strongly correlated with intractable focal epilepsy secondary to its cortical involvement. [9]
Immune etiologies of focal epilepsy should be considered when there is evidence of autoimmune-mediated central nervous system inflammation with recurrent seizures; in most cases, limbic and/or cortical involvement is seen in cases of focal immune-related epilepsy. While some antibodies have been implicated (see Table below), many remain undiscovered. Interestingly, some antibodies have been associated with particular semiologies; for instance, LGI-1 antibody encephalitis has been associated with faciobrachial dystonic seizures. An increasing recognition of the heterogeneity in autoimmune encephalitides has resulted in the proposal of various anatomical, serological, and etiological classification schemes. [10] As our knowledge of immunology expands, it is expected that further immune causes for focal epilepsy will be appreciated.
Up to one-third of focal epilepsies have no clearly defined etiology and are therefore categorized as “unknown” in the 2017 ILAE etiological classification. This may be either due to inadequate patient access to diagnostic resources or to microscopic lesions that elude contemporary diagnostic modalities. In many patients with normal MRIs who undergo epilepsy surgery, focal pathologies such as microgyria and gliosis are identified later histopathologically. [11]
Table. (Open Table in a new window)
| Etiology | Associated Conditions* |
|---|---|
| Structural | Malformations of cortical development (focal cortical dysplasias, lissencephaly, polymicrogyria, schizencephaly, hemimegalencephaly, hypothalamic hamartoma)
Vascular lesions (arteriovenous malformations, cerebral angiomas)
Hippocampal sclerosis
Hypoxic-ischemic lesions (ischemic and hemorrhagic strokes, cerebral venous sinus thrombosis, hypoxic-ischemic brain injury, porencephalic cysts)
Neoplasms (dysembryoplastic neuroepithelial tumors, gangliogliomas, metastatic tumors, etc.)
Traumatic brain injury Syndromic conditions (Sturge Weber syndrome, Tuberous Sclerosis, Rasmussen syndrome |
| Genetic | Tuberous sclerosis, Sturge Weber Syndrome, Autosomal-dominant nocturnal frontal lobe epilepsy, Autosomal-dominant lateral temporal lobe epilepsy, familial mesial temporal lobe epilepsy, familial focal epilepsy with variable foci Presumed genetic cause: benign focal epilepsies of childhood, epilepsy-aphasia spectrum |
| Infectious | Bacterial meningoencephalitis, viral encephalitis, neurocysticercosis, HIV, cytomegalovirus, toxoplasmosis, Lyme disease |
| Metabolic | Mitochondrial encephalopathy with lactic acidosis and stroke-like episodes, Alpers’ syndrome, peroxisomal disorders, pyridoxine-dependent epilepsy |
| Immune | Rasmussen syndrome, antineuronal nuclear antibody type 1 (anti-Hu) encephalitis, anti-N methyl-D-aspartate (anti-NMDA) receptor encephalitis, anti-leucine-rich glioma inactivated 1 (anti-LG1) encephalitis, dipeptidyl-peptidase-like protein 6 (DPPX-6) encephalitis, Contactin-associated protein-like 2 (CASPR2) encephalitis, Glutamic acid decarboxylase 65-antibody (GAD65) encephalitis |
| Unknown | This represents up to one-third of focal epilepsies |
*The example conditions provided are not exhaustive but represent some of the most common conditions associated with focal epilepsy. This list has been compiled based on the ILAE diagnostic manual available at www.epilepsydiagnosis.org.
Epidemiology
In a large 2017 meta-analysis, Fiest et al. reported that the lifetime prevalence of epilepsy was approximately 7.6 per 1000 (95% CI 6.17–9.38) in the United States and internationally. Low/middle-income countries have a statistically significant increased prevalence of epilepsy (8.75 per 1000, 95% CI 7.23–10.59) vs high-income countries (5.18 per 1000, 95% CI 3.75–7.15); this discrepancy is likely related to greater environmental risk factors and poorer access to healthcare in lower income countries. [12]
Approximately 60% of adult-onset epilepsies are focal epilepsies. Males are slightly more affected; however, this is thought to be related to females being more likely to conceal their epilepsy diagnosis. There is an age-related incidence pattern that follows a U-shaped curve, with a peak in the first year of life and an increase during the sixth and seventh decades. (See the image below.)
Over the last decade, the incidence of epilepsy in younger age groups has declined, perhaps due to improvements in perinatal care. Meanwhile, the incidence has increased in the elderly, perhaps owing to an improved life expectancy. [13]
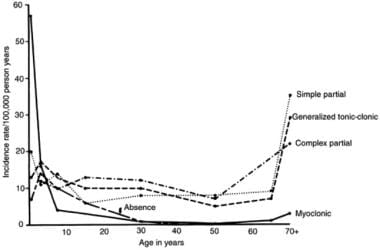 This graph illustrates the 2 peaks of incidence of epilepsy: early and late in life.
This graph illustrates the 2 peaks of incidence of epilepsy: early and late in life.
Prognosis
Although originally proposed in 1993, Sander’s prognostic classification for epilepsy patients continues to hold relevance today. [14] He divided patients into four categories as below:
Those with an excellent prognosis (about 20%–30% of total) and high probability of spontaneous remission (eg, benign focal epilepsies of childhood)
Those with a good prognosis (30%–40%) with easy pharmacological control and remission (some focal epilepsies)
Those with an uncertain prognosis (10%–20%) who may respond to drugs but tend to relapse upon withdrawal of treatment (most focal epilepsies)
Those with a poor prognosis (~20%) who tend to have intractable seizures despite intensive treatment (eg, progressive focal epilepsy syndromes)
Specific data are not available for focal epilepsies, however, epilepsy in general carries an increased risk of mortality; this may be secondary to sudden unexplained death in epilepsy (SUDEP), status epilepticus, accidental injuries, or suicide. The 2017 ILAE Mortality Task Force reported that epilepsy carried a standardized mortality ratio (observed deaths/expected deaths) of 2.3 in high-income countries and 2.6 in low-income countries. [15, 16]
In terms of quality of life, in 2016 epilepsy accounted for more than 13 million disability-adjusted life years (DALYs), a 20% decrease in comparison to 1990. [17] This likely represents advances in epilepsy diagnosis and treatment.
Patient Education
For patient education information, see the Brain and Nervous System Center, as well as Epilepsy.
-
This graph illustrates the 2 peaks of incidence of epilepsy: early and late in life.
Tables
| Etiology | Associated Conditions* |
|---|---|
| Structural | Malformations of cortical development (focal cortical dysplasias, lissencephaly, polymicrogyria, schizencephaly, hemimegalencephaly, hypothalamic hamartoma)
Vascular lesions (arteriovenous malformations, cerebral angiomas)
Hippocampal sclerosis
Hypoxic-ischemic lesions (ischemic and hemorrhagic strokes, cerebral venous sinus thrombosis, hypoxic-ischemic brain injury, porencephalic cysts)
Neoplasms (dysembryoplastic neuroepithelial tumors, gangliogliomas, metastatic tumors, etc.)
Traumatic brain injury Syndromic conditions (Sturge Weber syndrome, Tuberous Sclerosis, Rasmussen syndrome |
| Genetic | Tuberous sclerosis, Sturge Weber Syndrome, Autosomal-dominant nocturnal frontal lobe epilepsy, Autosomal-dominant lateral temporal lobe epilepsy, familial mesial temporal lobe epilepsy, familial focal epilepsy with variable foci Presumed genetic cause: benign focal epilepsies of childhood, epilepsy-aphasia spectrum |
| Infectious | Bacterial meningoencephalitis, viral encephalitis, neurocysticercosis, HIV, cytomegalovirus, toxoplasmosis, Lyme disease |
| Metabolic | Mitochondrial encephalopathy with lactic acidosis and stroke-like episodes, Alpers’ syndrome, peroxisomal disorders, pyridoxine-dependent epilepsy |
| Immune | Rasmussen syndrome, antineuronal nuclear antibody type 1 (anti-Hu) encephalitis, anti-N methyl-D-aspartate (anti-NMDA) receptor encephalitis, anti-leucine-rich glioma inactivated 1 (anti-LG1) encephalitis, dipeptidyl-peptidase-like protein 6 (DPPX-6) encephalitis, Contactin-associated protein-like 2 (CASPR2) encephalitis, Glutamic acid decarboxylase 65-antibody (GAD65) encephalitis |
| Unknown | This represents up to one-third of focal epilepsies |






